
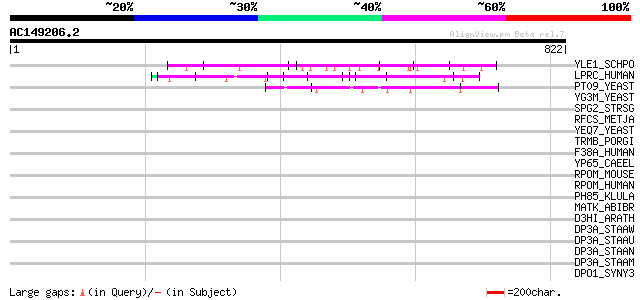
BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query= AC149206.2 - phase: 0
(822 letters)
Database: sprot
164,201 sequences; 59,974,054 total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
YLE1_SCHPO (Q10451) Hypothetical protein C1093.01 in chromosome I 70 2e-11
LPRC_HUMAN (P42704) 130 kDa leucine-rich protein (LRP 130) (GP13... 60 2e-08
PT09_YEAST (P32522) PET309 protein, mitochondrial precursor 52 6e-06
YG3M_YEAST (P48237) Hypothetical 101.4 kDa protein in RPL24B-RSR... 37 0.28
SPG2_STRSG (P19909) Immunoglobulin G binding protein G precursor... 36 0.36
RFCS_METJA (Q58817) Replication factor C small subunit (RFC smal... 35 0.81
YEQ7_YEAST (P40050) Hypothetical 79.5 kDa protein in PTP3-SER3 i... 35 1.1
TRMB_PORGI (Q7MVS9) tRNA (guanine-N(7)-)-methyltransferase (EC 2... 35 1.1
F38A_HUMAN (Q92508) Protein FAM38A 33 2.3
YP65_CAEEL (Q09214) Hypothetical protein B0495.5 in chromosome II 32 5.2
RPOM_MOUSE (Q8BKF1) DNA-directed RNA polymerase, mitochondrial p... 32 5.2
RPOM_HUMAN (O00411) DNA-directed RNA polymerase, mitochondrial p... 32 5.2
PH85_KLULA (Q92241) Negative regulator of the PHO system (EC 2.7... 32 5.2
MATK_ABIBR (Q8MDU0) Maturase K (Intron maturase) (Fragment) 32 5.2
D3HI_ARATH (Q9SUC0) Probable 3-hydroxyisobutyrate dehydrogenase,... 32 5.2
DP3A_STAAW (Q8NW58) DNA polymerase III alpha subunit (EC 2.7.7.7) 32 6.8
DP3A_STAAU (Q9F1K0) DNA polymerase III alpha subunit (EC 2.7.7.7) 32 6.8
DP3A_STAAN (P63980) DNA polymerase III alpha subunit (EC 2.7.7.7) 32 6.8
DP3A_STAAM (P63979) DNA polymerase III alpha subunit (EC 2.7.7.7) 32 6.8
DPO1_SYNY3 (Q55971) DNA polymerase I (EC 2.7.7.7) (POL I) 32 8.9
>YLE1_SCHPO (Q10451) Hypothetical protein C1093.01 in chromosome I
Length = 1261
Score = 70.1 bits (170), Expect = 2e-11
Identities = 74/322 (22%), Positives = 141/322 (42%), Gaps = 26/322 (8%)
Query: 425 SVAGGEIDVALMVRE--KMMEKGVFPDAQIYNVLMSGLCKKGRFPA-------AKLLLSE 475
S + ++DV ++ + K++E F ++Y L+S L K RF A +K L +
Sbjct: 780 SASPEQVDVNILFFQFGKLIETNKFLHPEVYPTLISVLSKNKRFDAVQRVFEHSKHLYRK 839
Query: 476 MLDLNLQPDAYMYATLVDGFIRNNELD---KATELF-EVVMSKGIDPGVVGYNVMIKGLC 531
+ +L+ + A ++D I ++ K++ LF + + G P + +I
Sbjct: 840 ISTKSLEKANWFMALILDAMILSSSFARQFKSSNLFCDNMKMLGYIPRASTFAHLINNST 899
Query: 532 KCGKMTDAVSYVN---KMKIANHAPDEYTHSTVIDGYVKQHDLDSALKMFGQMMKQKYKP 588
+ G DA + +N + K N P + ++ V+ + K+F +M + P
Sbjct: 900 RRGDTDDATTALNIFEETKRHNVKPSVFLYNAVLSKLGRARRTTECWKLFQEMKESGLLP 959
Query: 589 NVVAYTSLINGFCKIADMSRAEKVFRAMQSF-NLEPNVVTYTILIGGFSKT-GKPEKAAS 646
V Y ++IN C+I D S AEK+F M++ N +P V Y +I +T EKA
Sbjct: 960 TSVTYGTVINAACRIGDESLAEKLFAEMENQPNYQPRVAPYNTMIQFEVQTMFNREKALF 1019
Query: 647 FFELMLMNNCLPNDTTFHYLINGL-----TNITNTTLLIEKNEENDRSLILDFFAT---M 698
++ + + P+ T+ L++ N+ + ++E E D ++ +A +
Sbjct: 1020 YYNRLCATDIEPSSHTYKLLMDAYGTLKPVNVGSVKAVLELMERTDVPILSMHYAAYIHI 1079
Query: 699 ISEGWSQVIATYNSIIVCLCKH 720
+ S V A + + L KH
Sbjct: 1080 LGNVVSDVQAATSCYMNALAKH 1101
Score = 67.4 bits (163), Expect = 1e-10
Identities = 74/329 (22%), Positives = 138/329 (41%), Gaps = 19/329 (5%)
Query: 235 GCVPNVVFYNVIIDGYCKKGDLKRAT---RVFEELKLKGFLPTLETYGALIDGFCKAGKF 291
G +P + +I+ ++GD AT +FEE K P++ Y A++ +A +
Sbjct: 883 GYIPRASTFAHLINNSTRRGDTDDATTALNIFEETKRHNVKPSVFLYNAVLSKLGRARRT 942
Query: 292 QVVDQLLNEMNVMGLNVNVKVFNSIIDAKYKYGLVDKAAEMM-RMMTEMGCEPDITTYNI 350
+L EM GL + ++I+A + G A ++ M + +P + YN
Sbjct: 943 TECWKLFQEMKESGLLPTSVTYGTVINAACRIGDESLAEKLFAEMENQPNYQPRVAPYNT 1002
Query: 351 LINFSCSGGRIKE-AEEFLERAKERTLLPNKFSYTPLMHAYCKQGDYVMAS-DMLFKIAE 408
+I F +E A + R + P+ +Y LM AY + S + ++ E
Sbjct: 1003 MIQFEVQTMFNREKALFYYNRLCATDIEPSSHTYKLLMDAYGTLKPVNVGSVKAVLELME 1062
Query: 409 TGDKPDL-VSYGAFIH--GSVAGGEIDVALMVREKMMEK----GVFPDAQIYNVLMSGLC 461
D P L + Y A+IH G+V ++ A + K + DA ++ + L
Sbjct: 1063 RTDVPILSMHYAAYIHILGNVV-SDVQAATSCYMNALAKHDAGEIQLDANLFQSQIESLI 1121
Query: 462 KKGRFPAAKLLLSEMLDLNLQPDAYMYATLVDGFIRNNELDKATELFEVVMSKGI---DP 518
R ++S+M N+ +AY+ L+ GF + + KA F+++ +G+ +P
Sbjct: 1122 ANDRIVEGIQIVSDMKRYNVSLNAYIVNALIKGFTKVGMISKARYYFDLLECEGMSGKEP 1181
Query: 519 GVVGYNVMIKGLCKCGKMTDAVSYVNKMK 547
Y M++ A+ V ++K
Sbjct: 1182 ST--YENMVRAYLSVNDGRKAMEIVEQLK 1208
Score = 59.3 bits (142), Expect = 4e-08
Identities = 68/360 (18%), Positives = 140/360 (38%), Gaps = 49/360 (13%)
Query: 288 AGKFQVVDQLLNEMNVMGLNVNVKVFNSIIDAKYKYGLVDKAAEMMRMMTEM---GCEPD 344
A +F+ + + M ++G F +I+ + G D A + + E +P
Sbjct: 866 ARQFKSSNLFCDNMKMLGYIPRASTFAHLINNSTRRGDTDDATTALNIFEETKRHNVKPS 925
Query: 345 ITTYNILINFSCSGGRIKEAEEFLERAKERTLLPNKFSYTPLMHAYCKQGDYVMASDMLF 404
+ YN +++ R E + + KE LLP +Y +++A C+ GD +A +
Sbjct: 926 VFLYNAVLSKLGRARRTTECWKLFQEMKESGLLPTSVTYGTVINAACRIGDESLAEKLFA 985
Query: 405 KIA-ETGDKPDLVSYGAFIHGSV-AGGEIDVALMVREKMMEKGVFPDAQIYNVLMS--GL 460
++ + +P + Y I V + AL ++ + P + Y +LM G
Sbjct: 986 EMENQPNYQPRVAPYNTMIQFEVQTMFNREKALFYYNRLCATDIEPSSHTYKLLMDAYGT 1045
Query: 461 CKKGRFPAAK--LLLSEMLDL--------------------------------------N 480
K + K L L E D+
Sbjct: 1046 LKPVNVGSVKAVLELMERTDVPILSMHYAAYIHILGNVVSDVQAATSCYMNALAKHDAGE 1105
Query: 481 LQPDAYMYATLVDGFIRNNELDKATELFEVVMSKGIDPGVVGYNVMIKGLCKCGKMTDAV 540
+Q DA ++ + ++ I N+ + + ++ + + N +IKG K G ++ A
Sbjct: 1106 IQLDANLFQSQIESLIANDRIVEGIQIVSDMKRYNVSLNAYIVNALIKGFTKVGMISKAR 1165
Query: 541 SYVNKMKIANHAPDE-YTHSTVIDGYVKQHDLDSALKMFGQMMKQKYK-PNVVAYTSLIN 598
Y + ++ + E T+ ++ Y+ +D A+++ Q+ +++Y P V +SL+N
Sbjct: 1166 YYFDLLECEGMSGKEPSTYENMVRAYLSVNDGRKAMEIVEQLKRKRYPLPVVNRISSLVN 1225
Score = 53.9 bits (128), Expect = 2e-06
Identities = 65/286 (22%), Positives = 113/286 (38%), Gaps = 47/286 (16%)
Query: 413 PDLVSYGAFIHGSVAGGEID---VALMVREKMMEKGVFPDAQIYNVLMSGLCKKGRFPAA 469
P ++ I+ S G+ D AL + E+ V P +YN ++S L + R
Sbjct: 886 PRASTFAHLINNSTRRGDTDDATTALNIFEETKRHNVKPSVFLYNAVLSKLGRARRTTEC 945
Query: 470 KLLLSEMLDLNLQPDAYMYATLVDGFIRNNELDKATELF-EVVMSKGIDPGVVGYNVMIK 528
L EM + L P + Y T+++ R + A +LF E+ P V YN MI+
Sbjct: 946 WKLFQEMKESGLLPTSVTYGTVINAACRIGDESLAEKLFAEMENQPNYQPRVAPYNTMIQ 1005
Query: 529 -GLCKCGKMTDAVSYVNKMKIANHAPDEYTHSTVIDGYVKQHDLD-SALKMFGQMMKQKY 586
+ A+ Y N++ + P +T+ ++D Y ++ ++K ++M++
Sbjct: 1006 FEVQTMFNREKALFYYNRLCATDIEPSSHTYKLLMDAYGTLKPVNVGSVKAVLELMERTD 1065
Query: 587 KP--------------NVV-----AYTSLINGFCK--------------------IADMS 607
P NVV A + +N K IA+
Sbjct: 1066 VPILSMHYAAYIHILGNVVSDVQAATSCYMNALAKHDAGEIQLDANLFQSQIESLIANDR 1125
Query: 608 RAE--KVFRAMQSFNLEPNVVTYTILIGGFSKTGKPEKAASFFELM 651
E ++ M+ +N+ N LI GF+K G KA +F+L+
Sbjct: 1126 IVEGIQIVSDMKRYNVSLNAYIVNALIKGFTKVGMISKARYYFDLL 1171
>LPRC_HUMAN (P42704) 130 kDa leucine-rich protein (LRP 130) (GP130)
(Leucine-rich PPR-motif containing protein)
Length = 1273
Score = 60.1 bits (144), Expect = 2e-08
Identities = 38/160 (23%), Positives = 70/160 (43%), Gaps = 6/160 (3%)
Query: 504 ATELFEVVMSKGIDPGVVGYNVMIKGLCKCGKMTDAVSYVNKMKIANHAPDEYTHSTVID 563
A +++ + G V YN ++K + ++ KM+ AN P+ T+ +I
Sbjct: 25 AHRIWDTLQKLGAVYDVSHYNALLKVYLQNEYKFSPTDFLAKMEEANIQPNRVTYQRLIA 84
Query: 564 GYVKQHDLDSALKMFGQMMKQKYKPNVVAYTSLINGFCKIADMSRAEKVFRAMQSFNLEP 623
Y D++ A K+ G M + +++L+ G + DM AE + M+ +EP
Sbjct: 85 SYCNVGDIEGASKILGFMKTKDLPVTEAVFSALVTGHARAGDMENAENILTVMRDAGIEP 144
Query: 624 NVVTYTILIGGFSKTGKPE------KAASFFELMLMNNCL 657
TY L+ +++ G + + FEL LM+ L
Sbjct: 145 GPDTYLALLNAYAEKGDIDHVKQTLEKVEKFELHLMDRDL 184
Score = 58.9 bits (141), Expect = 5e-08
Identities = 44/190 (23%), Positives = 80/190 (41%), Gaps = 5/190 (2%)
Query: 220 KVEEGRKLIDDRWGN----GCVPNVVFYNVIIDGYCKKGDLKRATRVFEELKLKGFLPTL 275
K+EE + W G V +V YN ++ Y + T +++ P
Sbjct: 17 KLEERTEFAHRIWDTLQKLGAVYDVSHYNALLKVYLQNEYKFSPTDFLAKMEEANIQPNR 76
Query: 276 ETYGALIDGFCKAGKFQVVDQLLNEMNVMGLNVNVKVFNSIIDAKYKYGLVDKAAEMMRM 335
TY LI +C G + ++L M L V VF++++ + G ++ A ++ +
Sbjct: 77 VTYQRLIASYCNVGDIEGASKILGFMKTKDLPVTEAVFSALVTGHARAGDMENAENILTV 136
Query: 336 MTEMGCEPDITTYNILINFSCSGGRIKEAEEFLERAKERTLLPNKFSYTPLMHAYCKQGD 395
M + G EP TY L+N G I ++ LE+ ++ L ++ ++ K G
Sbjct: 137 MRDAGIEPGPDTYLALLNAYAEKGDIDHVKQTLEKVEKFELHLMDRDLLQIIFSFSKAG- 195
Query: 396 YVMASDMLFK 405
Y+ S +K
Sbjct: 196 YLSMSQKFWK 205
Score = 48.1 bits (113), Expect = 9e-05
Identities = 38/171 (22%), Positives = 78/171 (45%), Gaps = 8/171 (4%)
Query: 275 LETYGALIDGFCKAGKFQVVDQLLNEMNVMGLNVNVKVFNSIIDA----KYKYGLVDKAA 330
L + G+L+ + + ++ + + +G +V +N+++ +YK+ D A
Sbjct: 6 LRSCGSLLPELKLEERTEFAHRIWDTLQKLGAVYDVSHYNALLKVYLQNEYKFSPTDFLA 65
Query: 331 EMMRMMTEMGCEPDITTYNILINFSCSGGRIKEAEEFLERAKERTLLPNKFSYTPLMHAY 390
+M E +P+ TY LI C+ G I+ A + L K + L + ++ L+ +
Sbjct: 66 KM----EEANIQPNRVTYQRLIASYCNVGDIEGASKILGFMKTKDLPVTEAVFSALVTGH 121
Query: 391 CKQGDYVMASDMLFKIAETGDKPDLVSYGAFIHGSVAGGEIDVALMVREKM 441
+ GD A ++L + + G +P +Y A ++ G+ID EK+
Sbjct: 122 ARAGDMENAENILTVMRDAGIEPGPDTYLALLNAYAEKGDIDHVKQTLEKV 172
Score = 47.8 bits (112), Expect = 1e-04
Identities = 33/147 (22%), Positives = 65/147 (43%), Gaps = 8/147 (5%)
Query: 558 HSTVIDGYVKQHDLDSALKMFGQMMKQKYKPNVVAYTSLINGFCKIADMSRAEKVFRAMQ 617
++ ++ Y++ S +M + +PN V Y LI +C + D+ A K+ M+
Sbjct: 44 YNALLKVYLQNEYKFSPTDFLAKMEEANIQPNRVTYQRLIASYCNVGDIEGASKILGFMK 103
Query: 618 SFNLEPNVVTYTILIGGFSKTGKPEKAASFFELMLMNNCLPNDTTFHYLING------LT 671
+ +L ++ L+ G ++ G E A + +M P T+ L+N +
Sbjct: 104 TKDLPVTEAVFSALVTGHARAGDMENAENILTVMRDAGIEPGPDTYLALLNAYAEKGDID 163
Query: 672 NITNTTLLIEKNEEN--DRSLILDFFA 696
++ T +EK E + DR L+ F+
Sbjct: 164 HVKQTLEKVEKFELHLMDRDLLQIIFS 190
Score = 47.4 bits (111), Expect = 2e-04
Identities = 56/292 (19%), Positives = 111/292 (37%), Gaps = 23/292 (7%)
Query: 211 VVKGLCDVGKVEEGRKLIDDRWGNGCVPNVVFYNVIIDGYCKKGDLKRATRVFEELKLKG 270
++ C+VG +E K++ ++ ++ G+ + GD++ A + ++ G
Sbjct: 82 LIASYCNVGDIEGASKILGFMKTKDLPVTEAVFSALVTGHARAGDMENAENILTVMRDAG 141
Query: 271 FLPTLETYGALIDGFCKAGKFQVVDQLLNEMNVMGLNVNVKVFNSIIDAKYKYGLVDKAA 330
P +TY AL++ + + G V Q L ++ L++ + II + K G + +
Sbjct: 142 IEPGPDTYLALLNAYAEKGDIDHVKQTLEKVEKFELHLMDRDLLQIIFSFSKAGYLSMSQ 201
Query: 331 EMMRMMT-EMGCEPDITTYNILINFSCSGGRIKEAEEFLERAKERTLL---------PNK 380
+ + T E PD +L+ E LE + LL P+
Sbjct: 202 KFWKKFTCERRYIPDAMNLILLL-----------VTEKLEDVALQILLACPVSKEDGPSV 250
Query: 381 FSYTPLMHAYCKQGDYVMASDMLFKIAETGDKPDLVSYGAFIHGSVAGGEIDVALMVREK 440
F L H +D K+ E + + +H ++ + D+A + +
Sbjct: 251 FGSFFLQHCVTMNTPVEKLTDYCKKLKEVQMHSFPLQF--TLHCALLANKTDLAKALMKA 308
Query: 441 MMEKGVFPDAQIYNVLMSGLCKKGRFPAAKLLLSEMLDLNLQPDAYMYATLV 492
+ E+G + L+ G K+ +L M +L + PD Y V
Sbjct: 309 VKEEGFPIRPHYFWPLLVGRRKEKNVQGIIEILKGMQELGVHPDQETYTDYV 360
Score = 46.6 bits (109), Expect = 3e-04
Identities = 29/129 (22%), Positives = 57/129 (43%)
Query: 383 YTPLMHAYCKQGDYVMASDMLFKIAETGDKPDLVSYGAFIHGSVAGGEIDVALMVREKMM 442
Y L+ Y + +D L K+ E +P+ V+Y I G+I+ A + M
Sbjct: 44 YNALLKVYLQNEYKFSPTDFLAKMEEANIQPNRVTYQRLIASYCNVGDIEGASKILGFMK 103
Query: 443 EKGVFPDAQIYNVLMSGLCKKGRFPAAKLLLSEMLDLNLQPDAYMYATLVDGFIRNNELD 502
K + +++ L++G + G A+ +L+ M D ++P Y L++ + ++D
Sbjct: 104 TKDLPVTEAVFSALVTGHARAGDMENAENILTVMRDAGIEPGPDTYLALLNAYAEKGDID 163
Query: 503 KATELFEVV 511
+ E V
Sbjct: 164 HVKQTLEKV 172
Score = 43.5 bits (101), Expect = 0.002
Identities = 25/108 (23%), Positives = 47/108 (43%)
Query: 440 KMMEKGVFPDAQIYNVLMSGLCKKGRFPAAKLLLSEMLDLNLQPDAYMYATLVDGFIRNN 499
KM E + P+ Y L++ C G A +L M +L +++ LV G R
Sbjct: 66 KMEEANIQPNRVTYQRLIASYCNVGDIEGASKILGFMKTKDLPVTEAVFSALVTGHARAG 125
Query: 500 ELDKATELFEVVMSKGIDPGVVGYNVMIKGLCKCGKMTDAVSYVNKMK 547
+++ A + V+ GI+PG Y ++ + G + + K++
Sbjct: 126 DMENAENILTVMRDAGIEPGPDTYLALLNAYAEKGDIDHVKQTLEKVE 173
>PT09_YEAST (P32522) PET309 protein, mitochondrial precursor
Length = 965
Score = 52.0 bits (123), Expect = 6e-06
Identities = 60/293 (20%), Positives = 120/293 (40%), Gaps = 19/293 (6%)
Query: 447 FPDAQI----YNVLMSGLCKKGRFPAAKLLLSEMLDLNLQPDAYMYATLVDGFIRNNELD 502
+PDAQ ++ L+ + + A + + + + P Y L+ R ELD
Sbjct: 304 YPDAQHDQNQFDYLLVAHSRLHNWDALQQQFNALFGIGKLPSIQHYGILMYTMARIGELD 363
Query: 503 KATELFEVVMSKGIDPGVVGYNVMIKGLCKCGKMTDAVSYVNKMKIANHAPDEYTHSTVI 562
+L+ ++ +G+ P ++ K G S+ K + P TH+ ++
Sbjct: 364 SVNKLYTQLLRRGMIPTYAVLQSLLYAHYKVGDFAACFSHFELFKKYDITPSTATHTIML 423
Query: 563 DGYVKQHDLDSALKMFGQMMKQKYKPNVVA----YTSLINGFCKIADMSRAEKVFRAM-Q 617
Y +DLD A ++ ++ + P+V + LI CK + A+++F M +
Sbjct: 424 KVYRGLNDLDGAFRILKRLSED---PSVEITEGHFALLIQMCCKTTNHLIAQELFNLMTE 480
Query: 618 SFNLEPNVVTYTILIGGFSKTGKPEKAASFFELMLMNNCLPNDTTFHY-----LINGLTN 672
+N++ + + L+ + ++ +P +A + FE N + Y GL N
Sbjct: 481 HYNIQHTGKSISALMDVYIESNRPTEAIALFEKHSKNLSWRDGLISVYNKAIKAYIGLRN 540
Query: 673 ITNTTLLIEKNEENDRSLILDFFATMIS--EGWSQVIATYNSIIVCLCKHGMV 723
L +K + ++ +F+ MI ++ T SII L KH ++
Sbjct: 541 ANKCEELFDKITTSKLAVNSEFYKMMIKFLVTLNEDCETALSIIDQLIKHSVI 593
Score = 49.3 bits (116), Expect = 4e-05
Identities = 59/300 (19%), Positives = 123/300 (40%), Gaps = 19/300 (6%)
Query: 379 NKFSYTPLMHAYCKQGDYVMAS-DMLFKIAETGDKPDLVSYGAFIHGSVAGGEIDVALMV 437
N+F Y + H+ D + + LF I G P + YG ++ GE+D +
Sbjct: 312 NQFDYLLVAHSRLHNWDALQQQFNALFGI---GKLPSIQHYGILMYTMARIGELDSVNKL 368
Query: 438 REKMMEKGVFPDAQIYNVLMSGLCKKGRFPAAKLLLSEMLDLNLQPDAYMYATLVDGFIR 497
+++ +G+ P + L+ K G F A ++ P + ++ +
Sbjct: 369 YTQLLRRGMIPTYAVLQSLLYAHYKVGDFAACFSHFELFKKYDITPSTATHTIMLKVYRG 428
Query: 498 NNELDKATELFEVVMSKGIDPGVV----GYNVMIKGLCKCGKMTDAVSYVNKMKIANHAP 553
N+LD A F ++ DP V + ++I+ CK A N M H
Sbjct: 429 LNDLDGA---FRILKRLSEDPSVEITEGHFALLIQMCCKTTNHLIAQELFNLM--TEHYN 483
Query: 554 DEYTH---STVIDGYVKQHDLDSALKMFGQMMKQ-KYKPNVVA-YTSLINGFCKIADMSR 608
++T S ++D Y++ + A+ +F + K ++ +++ Y I + + + ++
Sbjct: 484 IQHTGKSISALMDVYIESNRPTEAIALFEKHSKNLSWRDGLISVYNKAIKAYIGLRNANK 543
Query: 609 AEKVFRAMQSFNLEPNVVTYTILIGGFSKTGKP-EKAASFFELMLMNNCLPNDTTFHYLI 667
E++F + + L N Y ++I + E A S + ++ ++ + D T +I
Sbjct: 544 CEELFDKITTSKLAVNSEFYKMMIKFLVTLNEDCETALSIIDQLIKHSVIKVDATHFEII 603
Score = 42.4 bits (98), Expect = 0.005
Identities = 55/268 (20%), Positives = 105/268 (38%), Gaps = 17/268 (6%)
Query: 327 DKAAEMMRMMTEMGCEPDITTYNILINFSCSGGRIKEAEEFLERAKERTLLPNKFSYTPL 386
D + + +G P I Y IL+ G + + + R ++P L
Sbjct: 328 DALQQQFNALFGIGKLPSIQHYGILMYTMARIGELDSVNKLYTQLLRRGMIPTYAVLQSL 387
Query: 387 MHAYCKQGDYVMASDMLFKIAETGDKPDLVSYGAFIHGSVAGGEIDVALMVREKMMEKGV 446
++A+ K GD+ + P ++ + ++D A + +++ E
Sbjct: 388 LYAHYKVGDFAACFSHFELFKKYDITPSTATHTIMLKVYRGLNDLDGAFRILKRLSED-- 445
Query: 447 FPDAQI----YNVLMSGLCKKGRFPAAKLLLSEMLD-LNLQPDAYMYATLVDGFIRNNEL 501
P +I + +L+ CK A+ L + M + N+Q + L+D +I +N
Sbjct: 446 -PSVEITEGHFALLIQMCCKTTNHLIAQELFNLMTEHYNIQHTGKSISALMDVYIESNRP 504
Query: 502 DKATELFEVVMSKGI---DPGVVGYNVMIK---GLCKCGKMTDAVSYVNKMKIANHAPDE 555
+A LFE SK + D + YN IK GL K + + K+A ++ E
Sbjct: 505 TEAIALFE-KHSKNLSWRDGLISVYNKAIKAYIGLRNANKCEELFDKITTSKLAVNS--E 561
Query: 556 YTHSTVIDGYVKQHDLDSALKMFGQMMK 583
+ + D ++AL + Q++K
Sbjct: 562 FYKMMIKFLVTLNEDCETALSIIDQLIK 589
Score = 40.0 bits (92), Expect = 0.025
Identities = 59/329 (17%), Positives = 121/329 (35%), Gaps = 47/329 (14%)
Query: 270 GFLPTLETYGALIDGFCKAGKFQVVDQLLNEMNVMGLNVNVKVFNSIIDAKYKYGLVDKA 329
G LP+++ YG L+ + G+ V++L ++ G+ V S++ A YK G
Sbjct: 341 GKLPSIQHYGILMYTMARIGELDSVNKLYTQLLRRGMIPTYAVLQSLLYAHYKVGDFAAC 400
Query: 330 AEMMRMMTEMGCEPDITTYNILINFSCSGGRIKEAEEFLERAKERTLLP-NKFSYTPLMH 388
+ + P T+ I++ + A L+R E + + + L+
Sbjct: 401 FSHFELFKKYDITPSTATHTIMLKVYRGLNDLDGAFRILKRLSEDPSVEITEGHFALLIQ 460
Query: 389 AYCKQGDYVMASDML------FKIAETGDKPDLVSYGAFIHGSVAGGEIDVALMVREKMM 442
CK ++++A ++ + I TG S A + + A+ + EK
Sbjct: 461 MCCKTTNHLIAQELFNLMTEHYNIQHTGK-----SISALMDVYIESNRPTEAIALFEKHS 515
Query: 443 EKGVFPDAQIYNVLMSGLCKKGRFPAAKLLLSEMLDLNLQPDAYMYATLVDGFIRNNELD 502
+ + D I +Y + +I +
Sbjct: 516 KNLSWRDGLI---------------------------------SVYNKAIKAYIGLRNAN 542
Query: 503 KATELFEVVMSKGIDPGVVGYNVMIKGLCKCGKMTD-AVSYVNKMKIANHAPDEYTHSTV 561
K ELF+ + + + Y +MIK L + + A+S ++++ + + TH +
Sbjct: 543 KCEELFDKITTSKLAVNSEFYKMMIKFLVTLNEDCETALSIIDQLIKHSVIKVDATHFEI 602
Query: 562 I-DGYVKQHDLDSALKMFGQMMKQKYKPN 589
I + Y K+ D + ++ M + K N
Sbjct: 603 IMEAYDKEGYRDGIINLYKTMSQNKVPAN 631
Score = 32.7 bits (73), Expect = 4.0
Identities = 14/60 (23%), Positives = 31/60 (51%)
Query: 232 WGNGCVPNVVFYNVIIDGYCKKGDLKRATRVFEELKLKGFLPTLETYGALIDGFCKAGKF 291
+G G +P++ Y +++ + G+L +++ +L +G +PT +L+ K G F
Sbjct: 338 FGIGKLPSIQHYGILMYTMARIGELDSVNKLYTQLLRRGMIPTYAVLQSLLYAHYKVGDF 397
>YG3M_YEAST (P48237) Hypothetical 101.4 kDa protein in RPL24B-RSR1
intergenic region
Length = 864
Score = 36.6 bits (83), Expect = 0.28
Identities = 25/111 (22%), Positives = 50/111 (44%), Gaps = 3/111 (2%)
Query: 524 NVMIKGLCKCGKMTDAVSYVNKMKIA-NHAPDEYTHSTVIDGYVKQHDLDSALKMFGQM- 581
N +K KC D ++ K + P++ +TVI Y ++ A F M
Sbjct: 288 NNCLKYSTKCSSFHDMDYFITKFRDDYGITPNKQNLTTVIQFYSRKEMTKQAWNTFDTMK 347
Query: 582 -MKQKYKPNVVAYTSLINGFCKIADMSRAEKVFRAMQSFNLEPNVVTYTIL 631
+ K+ P++ Y +++ K + +A +F+ +Q N++P TY ++
Sbjct: 348 FLSTKHFPDICTYNTMLRICEKERNFPKALDLFQEIQDHNIKPTTNTYIMM 398
>SPG2_STRSG (P19909) Immunoglobulin G binding protein G precursor
(IgG binding protein G)
Length = 593
Score = 36.2 bits (82), Expect = 0.36
Identities = 48/201 (23%), Positives = 73/201 (35%), Gaps = 33/201 (16%)
Query: 448 PDAQIYNVLMSGLCKKGRFPAAKLLLSEMLDLNLQPDAYMYATLVDGFIRNNELDKATEL 507
P Y ++++G KG +E +D + +G D AT+
Sbjct: 299 PKTDTYKLILNGKTLKGE------TTTEAVDAATAEKVFKQYANDNGVDGEWTYDDATKT 352
Query: 508 F------EVVMSKGIDPGVVGYNVMIKGLCKCGK-MTDAVSYVNKMKIANHAPDEYTHST 560
F EV+ + + P V Y ++I G G+ T+AV K+ +Y +
Sbjct: 353 FTVTEKPEVIDASELTPAVTTYKLVINGKTLKGETTTEAVDAATAEKVFK----QYANDN 408
Query: 561 VIDGYVKQHDLDSALKMFGQMMK------QKYKPNVVAYTSLING-------FCKIADMS 607
+DG + D A K F K + P V Y +ING K D
Sbjct: 409 GVDG---EWTYDDATKTFTVTEKPEVIDASELTPAVTTYKLVINGKTLKGETTTKAVDAE 465
Query: 608 RAEKVFRAMQSFNLEPNVVTY 628
AEK F+ + N V TY
Sbjct: 466 TAEKAFKQYANDNGVDGVWTY 486
Score = 34.7 bits (78), Expect = 1.1
Identities = 44/169 (26%), Positives = 66/169 (39%), Gaps = 28/169 (16%)
Query: 523 YNVMIKGLCKCGKMT-DAVSYVNKMKIANHAPDEYTHSTVIDGYVKQHDLDSALKMFGQM 581
Y +++ G G+ T +AV K+ +Y + +DG + D A K F
Sbjct: 304 YKLILNGKTLKGETTTEAVDAATAEKVFK----QYANDNGVDG---EWTYDDATKTFTVT 356
Query: 582 MK------QKYKPNVVAYTSLING-------FCKIADMSRAEKVFRAMQSFNLEPNVVTY 628
K + P V Y +ING + D + AEKVF+ + N TY
Sbjct: 357 EKPEVIDASELTPAVTTYKLVINGKTLKGETTTEAVDAATAEKVFKQYANDNGVDGEWTY 416
Query: 629 TILIGGFSKTGKPEKAASFFELMLMNNCLPNDTTFHYLINGLTNITNTT 677
F+ T KP E++ + P TT+ +ING T TT
Sbjct: 417 DDATKTFTVTEKP-------EVIDASELTPAVTTYKLVINGKTLKGETT 458
>RFCS_METJA (Q58817) Replication factor C small subunit (RFC small
subunit) (Clamp loader small subunit) [Contains: Mja
RFC-1 intein; Mja RFC-2 intein; Mja RFC-3 intein]
Length = 1847
Score = 35.0 bits (79), Expect = 0.81
Identities = 30/99 (30%), Positives = 51/99 (51%), Gaps = 10/99 (10%)
Query: 289 GKFQVVDQLLNEMNVMGLNVNVKVFNSIIDAKYKYGLVDKAAEMMRMMTEMGCEPDITTY 348
GKF V L N + V+G++ + K+ + YK DK ++++ T+MG E +TTY
Sbjct: 80 GKFGVT--LTNNLKVLGIDEDGKIREFDVQYVYK----DKTNTLIKIKTKMGRELKVTTY 133
Query: 349 N-ILINFSCSGGRIKEAEEFL---ERAKERTLLPNKFSY 383
+ +LIN + ++AE + A R +L N+ Y
Sbjct: 134 HPLLINHKNGEIKWEKAENLKVGDKLATPRYILFNESDY 172
>YEQ7_YEAST (P40050) Hypothetical 79.5 kDa protein in PTP3-SER3
intergenic region
Length = 688
Score = 34.7 bits (78), Expect = 1.1
Identities = 36/157 (22%), Positives = 71/157 (44%), Gaps = 15/157 (9%)
Query: 484 DAYMYATLVDGFIRNNELDKATELFEVVMSKGIDPGVVGYNVMIKGLCK-CGKMTDAVSY 542
D+ + A V F++ +L+KA L + K GVVG N+++K + AV
Sbjct: 166 DSKLIANDVQKFLKRGQLEKAVFLARLAKKK----GVVGMNLIMKYYIEVVQSQQSAVDI 221
Query: 543 VNKMKIANHAPDEYTHSTVIDGYVKQHDLDSALKMFGQMMKQ-------KYKPNVVAYTS 595
N K D+++ + + +G KQ +L S K +G+++ + K + + Y +
Sbjct: 222 FNWRKKWGVPIDQHSITILFNGLSKQENLVS--KKYGELVLKTIDSLCDKNELTEIEYNT 279
Query: 596 LINGFCKIADMSRAEKVFRAMQSFNLEPNVVTYTILI 632
+ D + K+ + L+ + +TYT++I
Sbjct: 280 ALAALINCTDETLVFKLLN-KKCPGLKKDSITYTLMI 315
>TRMB_PORGI (Q7MVS9) tRNA (guanine-N(7)-)-methyltransferase (EC
2.1.1.33) (tRNA(m7G46)-methyltransferase)
Length = 244
Score = 34.7 bits (78), Expect = 1.1
Identities = 20/56 (35%), Positives = 31/56 (54%), Gaps = 3/56 (5%)
Query: 186 LYDKMLERGGDHGLDLVVDNYSIVIVVKGLCDVGKVEEGRKLIDDRWGNGCVPNVV 241
LYDK+LERGG + L D+ + K L ++ + ++ DD +G GCV N +
Sbjct: 134 LYDKVLERGG--RIHLKTDSPFLYTYTKALVELNGLPV-HEITDDLYGKGCVENEI 186
>F38A_HUMAN (Q92508) Protein FAM38A
Length = 2035
Score = 33.5 bits (75), Expect = 2.3
Identities = 31/110 (28%), Positives = 47/110 (42%), Gaps = 10/110 (9%)
Query: 645 ASFFELMLMNNCLPNDTTFHYLINGLTNITNTTLLIEKNEENDRSLILDFFATMISEGWS 704
A F+ L+ L DT ++ + N T++I KN SL+ F + G+
Sbjct: 706 ACFYLLLFGTALLQRDTRARLVLWDCLILYNVTVIISKNM---LSLLACVFVEQMQTGFC 762
Query: 705 QVIATYNSIIVCLCK-----HGMVDTAQLLQTKMLRKGFLMDSVCFSALL 749
VI ++ +VC K M+D Q + G + DSVCF LL
Sbjct: 763 WVIQLFS--LVCTVKGYYDPKEMMDRDQDCLLPVEEAGIIWDSVCFFFLL 810
>YP65_CAEEL (Q09214) Hypothetical protein B0495.5 in chromosome II
Length = 729
Score = 32.3 bits (72), Expect = 5.2
Identities = 19/70 (27%), Positives = 32/70 (45%)
Query: 25 PHIKTLIHDVIQILKTNQSHHSLQSRFAESQIIVSNVAHFVIDRIHNPQHGLYFFHWAST 84
PH + +++D Q+L T H L R ++ V N + + +I + G Y A +
Sbjct: 280 PHFEKMLYDQSQLLATYSDFHKLTERKHDNVKHVINDIYQYMQKISHKDGGFYAAEDADS 339
Query: 85 LPFSSPLNNV 94
LP + N V
Sbjct: 340 LPNHNSSNKV 349
>RPOM_MOUSE (Q8BKF1) DNA-directed RNA polymerase, mitochondrial
precursor (EC 2.7.7.6) (MtRPOL)
Length = 1207
Score = 32.3 bits (72), Expect = 5.2
Identities = 31/154 (20%), Positives = 62/154 (40%), Gaps = 5/154 (3%)
Query: 420 AFIHGSVAGGEIDVA---LMVREKMMEKGVFPDAQIYNVLMSGLCKKGRFPAAKLLLSEM 476
AF V G++ +A L+ ++ +YN +M G +KG F + +
Sbjct: 200 AFFECCVCTGQVPLAHHVLVTHHNNGDRQQVLTLHMYNTVMLGWARKGSFRELVYVFLML 259
Query: 477 LDLNLQPDAYMYATLVDGF-IRNNELDKATELFEVVMSKGIDPGVVGYNVMIKGLCKCGK 535
D L PD YA + R+ ++ + +M +G P ++ +++++ +
Sbjct: 260 KDAGLSPDLCSYAAALQCMGRRDQDVRTIQRCLKQMMEEGFQPQLLFTDLVLEEEDRAAL 319
Query: 536 MTDAVSYVNKMKIANHAPDEYTHSTVI-DGYVKQ 568
+ V + AP ST++ D Y K+
Sbjct: 320 LRAVVKAEPAFRPPPQAPSPVNTSTLLKDIYSKE 353
>RPOM_HUMAN (O00411) DNA-directed RNA polymerase, mitochondrial
precursor (EC 2.7.7.6) (MtRPOL)
Length = 1230
Score = 32.3 bits (72), Expect = 5.2
Identities = 27/124 (21%), Positives = 56/124 (44%), Gaps = 8/124 (6%)
Query: 243 YNVIIDGYCKKGDLKRATRVFEELKLKGFLPTLETYGALIDGFCKAGKFQ---VVDQLLN 299
YN ++ G+ ++G K V +K G P L +Y A + C + Q +++ L
Sbjct: 264 YNAVMLGWARQGAFKELVYVLFMVKDAGLTPDLLSYAAALQ--CMGRQDQDAGTIERCLE 321
Query: 300 EMNVMGLNVNVKVFNSIIDAKYKYGLVDKAAEMMRMMTEMGCE--PDITTYNILINFSCS 357
+M+ GL + +F +++ ++ V KA ++ + + P + T +L +
Sbjct: 322 QMSQEGLKLQA-LFTAVLLSEEDRATVLKAVHKVKPTFSLPPQLPPPVNTSKLLRDVYAK 380
Query: 358 GGRI 361
GR+
Sbjct: 381 DGRV 384
>PH85_KLULA (Q92241) Negative regulator of the PHO system (EC
2.7.1.37) (Serine/threonine-protein kinase PHO85)
Length = 304
Score = 32.3 bits (72), Expect = 5.2
Identities = 28/74 (37%), Positives = 35/74 (46%), Gaps = 9/74 (12%)
Query: 683 NEENDRSLILDFFATMISEGWSQV--IATYNSIIVCLCKHGMVDTAQLLQTKMLRKGFLM 740
N+E LI D T + + W QV +A YN + L H D QLLQ + L
Sbjct: 213 NDEEQLKLIFDTMGTPVEQTWPQVTQLAKYNPL---LPPHMPRDLKQLLQNN--TEEVLD 267
Query: 741 DSVCFSALLHGLCQ 754
D+V LLHGL Q
Sbjct: 268 DNVV--DLLHGLLQ 279
>MATK_ABIBR (Q8MDU0) Maturase K (Intron maturase) (Fragment)
Length = 508
Score = 32.3 bits (72), Expect = 5.2
Identities = 31/119 (26%), Positives = 50/119 (41%), Gaps = 17/119 (14%)
Query: 5 ILSLIKPHHHPKPSSSSSLPPHIKTLIHDVIQILKTNQSHHSLQS------------RFA 52
++ L+K HP+ SS S P +T I+ + N +SL+S R+A
Sbjct: 221 LVPLLKQSFHPRSSSHGSFPD--RTHFDRKIKHIIRNSRRNSLKSIWSLKDPRIHYVRYA 278
Query: 53 ESQIIVSNVAHFVID--RIHNPQHGLYFFH-WASTLPFSSPLNNVAYSSLLKLMVKYRL 108
E II H ++ R H P ++FH W+ S + SS L ++ R+
Sbjct: 279 ERSIIAIKGTHLLVKKCRYHLPIFRQFYFHLWSEPYRVCSHQLSKNCSSSLGYFLRVRM 337
>D3HI_ARATH (Q9SUC0) Probable 3-hydroxyisobutyrate dehydrogenase,
mitochondrial precursor (EC 1.1.1.31) (HIBADH)
Length = 347
Score = 32.3 bits (72), Expect = 5.2
Identities = 19/65 (29%), Positives = 32/65 (49%), Gaps = 4/65 (6%)
Query: 290 KFQVVDQLLNEMNVMGL----NVNVKVFNSIIDAKYKYGLVDKAAEMMRMMTEMGCEPDI 345
+F Q N+ +G N+ ++ N++I A YK + D ++M+M TEMG
Sbjct: 26 RFSSSSQNSNQFQNVGFIGLGNMGFRMVNNLIRAGYKVTVHDINRDVMKMFTEMGVSSRE 85
Query: 346 TTYNI 350
T Y +
Sbjct: 86 TPYEV 90
>DP3A_STAAW (Q8NW58) DNA polymerase III alpha subunit (EC 2.7.7.7)
Length = 1065
Score = 32.0 bits (71), Expect = 6.8
Identities = 24/101 (23%), Positives = 45/101 (43%), Gaps = 11/101 (10%)
Query: 590 VVAYTSLINGFCKIADMSRAEKVFRAMQSFNLEPNVVTYTILIGGFSKTGKPEKAASFFE 649
+VAY ++ + + + E R S N++ +T T ++ GF K F++
Sbjct: 1 MVAYLNIHTAYDLLNSSLKIEDAVRLAVSENVDALAITDTNVLYGFPK---------FYD 51
Query: 650 LMLMNNCLPNDTTFHYLINGLTNITNTTLLIEKNEENDRSL 690
+ NN P Y+ NGL + T+++ KN + + L
Sbjct: 52 ACIANNIKPIFGMTIYVTNGLNTV--ETVVLAKNNDGLKDL 90
>DP3A_STAAU (Q9F1K0) DNA polymerase III alpha subunit (EC 2.7.7.7)
Length = 1065
Score = 32.0 bits (71), Expect = 6.8
Identities = 24/101 (23%), Positives = 45/101 (43%), Gaps = 11/101 (10%)
Query: 590 VVAYTSLINGFCKIADMSRAEKVFRAMQSFNLEPNVVTYTILIGGFSKTGKPEKAASFFE 649
+VAY ++ + + + E R S N++ +T T ++ GF K F++
Sbjct: 1 MVAYLNIHTAYDLLNSSLKIEDAVRLAVSENVDALAITDTNVLYGFPK---------FYD 51
Query: 650 LMLMNNCLPNDTTFHYLINGLTNITNTTLLIEKNEENDRSL 690
+ NN P Y+ NGL + T+++ KN + + L
Sbjct: 52 ACIANNIKPIFGMTIYVTNGLNTV--ETVVLAKNNDGLKDL 90
>DP3A_STAAN (P63980) DNA polymerase III alpha subunit (EC 2.7.7.7)
Length = 1065
Score = 32.0 bits (71), Expect = 6.8
Identities = 24/101 (23%), Positives = 45/101 (43%), Gaps = 11/101 (10%)
Query: 590 VVAYTSLINGFCKIADMSRAEKVFRAMQSFNLEPNVVTYTILIGGFSKTGKPEKAASFFE 649
+VAY ++ + + + E R S N++ +T T ++ GF K F++
Sbjct: 1 MVAYLNIHTAYDLLNSSLKIEDAVRLAVSENVDALAITDTNVLYGFPK---------FYD 51
Query: 650 LMLMNNCLPNDTTFHYLINGLTNITNTTLLIEKNEENDRSL 690
+ NN P Y+ NGL + T+++ KN + + L
Sbjct: 52 ACIANNIKPIFGMTIYVTNGLNTV--ETVVLAKNNDGLKDL 90
>DP3A_STAAM (P63979) DNA polymerase III alpha subunit (EC 2.7.7.7)
Length = 1065
Score = 32.0 bits (71), Expect = 6.8
Identities = 24/101 (23%), Positives = 45/101 (43%), Gaps = 11/101 (10%)
Query: 590 VVAYTSLINGFCKIADMSRAEKVFRAMQSFNLEPNVVTYTILIGGFSKTGKPEKAASFFE 649
+VAY ++ + + + E R S N++ +T T ++ GF K F++
Sbjct: 1 MVAYLNIHTAYDLLNSSLKIEDAVRLAVSENVDALAITDTNVLYGFPK---------FYD 51
Query: 650 LMLMNNCLPNDTTFHYLINGLTNITNTTLLIEKNEENDRSL 690
+ NN P Y+ NGL + T+++ KN + + L
Sbjct: 52 ACIANNIKPIFGMTIYVTNGLNTV--ETVVLAKNNDGLKDL 90
>DPO1_SYNY3 (Q55971) DNA polymerase I (EC 2.7.7.7) (POL I)
Length = 986
Score = 31.6 bits (70), Expect = 8.9
Identities = 36/150 (24%), Positives = 63/150 (42%), Gaps = 21/150 (14%)
Query: 378 PNKFSYTPLMHAYCKQGDYVMASDMLFKIAETGDKPDLVSYGAFIHGSVAGGEIDVALMV 437
PN+ +Y PL H +Q + D+L +I P ++ F +A I++ +V
Sbjct: 425 PNQVAYIPLKHHQGEQLSLGIIKDLLGEILGNAIYPKVLQNAKFDRRVLAHHGIELGGVV 484
Query: 438 REKMMEKGVFPDAQIYNVLMSGLCKKGRFPAAKLLLSEMLDLNLQPDAYM---------- 487
+ M+ V + +N ++ LC+ R+ + LS DL L+ D +
Sbjct: 485 LDTMLASYVLQPEETHN--LTDLCR--RYNLGLVALS-YKDLGLKKDQTIADLPLETAGQ 539
Query: 488 ------YATLVDGFIRNNELDKATELFEVV 511
YAT + ELD+ EL E++
Sbjct: 540 YCGLDCYATYLLASKLQKELDQYPELKEIL 569
Database: sprot
Posted date: Nov 25, 2004 10:54 AM
Number of letters in database: 59,974,054
Number of sequences in database: 164,201
Lambda K H
0.322 0.138 0.406
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 94,314,041
Number of Sequences: 164201
Number of extensions: 4005386
Number of successful extensions: 9933
Number of sequences better than 10.0: 20
Number of HSP's better than 10.0 without gapping: 5
Number of HSP's successfully gapped in prelim test: 15
Number of HSP's that attempted gapping in prelim test: 9867
Number of HSP's gapped (non-prelim): 64
length of query: 822
length of database: 59,974,054
effective HSP length: 119
effective length of query: 703
effective length of database: 40,434,135
effective search space: 28425196905
effective search space used: 28425196905
T: 11
A: 40
X1: 16 ( 7.4 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 41 (21.9 bits)
S2: 70 (31.6 bits)
Medicago: description of AC149206.2