
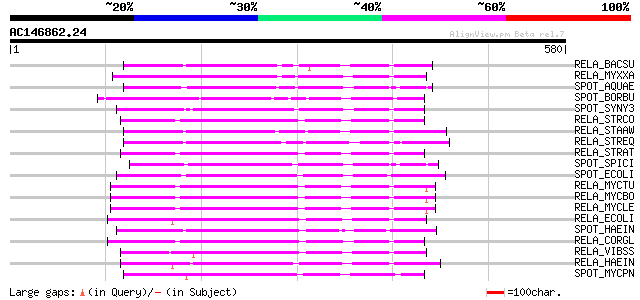
BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query= AC146862.24 - phase: 0
(580 letters)
Database: sprot
164,201 sequences; 59,974,054 total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
RELA_BACSU (O54408) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP ... 157 9e-38
RELA_MYXXA (O52177) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP ... 156 2e-37
SPOT_AQUAE (O67012) Guanosine-3',5'-bis(diphosphate) 3'-pyrophos... 154 6e-37
SPOT_BORBU (O51216) Guanosine-3',5'-bis(diphosphate) 3'-pyrophos... 153 1e-36
SPOT_SYNY3 (P74007) Probable guanosine-3',5'-bis(diphosphate) 3'... 151 4e-36
RELA_STRCO (P52560) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP ... 148 3e-35
RELA_STAAW (O32419) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP ... 147 7e-35
RELA_STREQ (Q54089) Putative GTP pyrophosphokinase (EC 2.7.6.5) ... 146 2e-34
RELA_STRAT (O85709) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP ... 145 2e-34
SPOT_SPICI (O34098) Guanosine-3',5'-bis(diphosphate) 3'-pyrophos... 145 4e-34
SPOT_ECOLI (P17580) Guanosine-3',5'-bis(diphosphate) 3'-pyrophos... 140 7e-33
RELA_MYCTU (P66014) Probable GTP pyrophosphokinase (EC 2.7.6.5) ... 135 3e-31
RELA_MYCBO (P66015) Probable GTP pyrophosphokinase (EC 2.7.6.5) ... 135 3e-31
RELA_MYCLE (Q49640) Probable GTP pyrophosphokinase (EC 2.7.6.5) ... 135 4e-31
RELA_ECOLI (P11585) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP ... 128 4e-29
SPOT_HAEIN (P43811) Guanosine-3',5'-bis(diphosphate) 3'-pyrophos... 128 5e-29
RELA_CORGL (O87331) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP ... 125 4e-28
RELA_VIBSS (P55133) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP ... 119 2e-26
RELA_HAEIN (P44644) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP ... 118 5e-26
SPOT_MYCPN (P75386) Probable guanosine-3',5'-bis(diphosphate) 3'... 108 5e-23
>RELA_BACSU (O54408) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP
3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 734
Score = 157 bits (396), Expect = 9e-38
Identities = 103/328 (31%), Positives = 174/328 (52%), Gaps = 31/328 (9%)
Query: 120 LSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESLRVKNFASR 179
+ +A +L DL+MD I+ G L +V+E ++ L +++ A L+ ++ +
Sbjct: 55 IQVAGILVDLEMDPSTIAGGFLHDVVEDTDVTLDDLKEAFSEEVAMLVDGVTKLGKIKYK 114
Query: 180 VDILDDENAAALRK-FCLTYYDIRALILDLALKLDMMRHLGHLPRYQQQIISLQVMKIYA 238
+++ A RK F DIR +++ LA +L MR L HLP+ +Q+ IS + ++I+A
Sbjct: 115 SQ--EEQQAENHRKMFVAMAQDIRVILIKLADRLHNMRTLKHLPQEKQRRISNETLEIFA 172
Query: 239 PLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKDELLESLKSD 298
PLAH +G + I ELED + +YL P Y + ++ + ++Y DE++ +K
Sbjct: 173 PLAHRLGISKIKWELEDTALRYLNPQQYYRIVNLMKKKRAER----ELYVDEVVNEVK-- 226
Query: 299 PILAELVDDISVK----GRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSRENALE 354
+ V+++++K GR K YS +K++ ++ ++ D+L +R+++N
Sbjct: 227 ----KRVEEVNIKADFSGRPKHIYSIYRKMVLQNKQFNEIYDLLAVRILVN--------- 273
Query: 355 AGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLMEIQIR 414
+ CY II + WK +P R KDYI+ PK N Y+SLH V IG +E+QIR
Sbjct: 274 -SIKDCYAVLGIIHTCWKPMPGRFKDYIAMPKPNMYQSLHTTV----IGPKADPLEVQIR 328
Query: 415 TTEMDRLAVGGMASHSLYKAGLTNPEEA 442
T EM +A G+A+H YK G E A
Sbjct: 329 TFEMHEIAEYGVAAHWAYKEGKAANEGA 356
>RELA_MYXXA (O52177) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP
3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 757
Score = 156 bits (394), Expect = 2e-37
Identities = 102/330 (30%), Positives = 168/330 (50%), Gaps = 22/330 (6%)
Query: 108 LTPDGRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLL 167
L G L L +A +L +L++D I G+L + +E E+ GS AHL+
Sbjct: 56 LRKSGEPYLVHPLEVAGILGELKLDEASIVTGLLHDTIEDTLATEEELTELFGSEVAHLV 115
Query: 168 HESLRVKNFASRVDILDDENAAA-LRKFCLTY-YDIRALILDLALKLDMMRHLGHLPRYQ 225
++ F++ + +E A RK + DIR +++ LA + MR L H+ +
Sbjct: 116 DGVTKLSKFSASASLSQEEKQAENFRKMIIAMAQDIRVILVKLADRTHNMRTLDHMSEEK 175
Query: 226 QQIISLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLID 285
Q I+ + + IYAPLA+ +G ++I ELEDLSF+Y+ P + + L ++ +
Sbjct: 176 QARIAQETLDIYAPLANRLGISWIKTELEDLSFRYVKPQEFFALQAKLNKRKKER----E 231
Query: 286 VYKDELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLN 345
Y ++ + ++S LAE V GR+K YS KK+ G + ++D++ R++
Sbjct: 232 KYIEDTCDLIRSK--LAERGLKGEVSGRFKHVYSIYKKIKSQGIDFDQIHDIIAFRIIAP 289
Query: 346 PKSRENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRT 405
+CY A ++ MWK +P R KD+I+ PK N Y+SLH + IG
Sbjct: 290 TAP----------SCYEALGLVHEMWKPVPGRFKDFIAIPKPNMYQSLHTTI----IGPL 335
Query: 406 RPLMEIQIRTTEMDRLAVGGMASHSLYKAG 435
+E+QIRT+EM ++A G+A+H YK G
Sbjct: 336 SERVEVQIRTSEMHKIAEEGIAAHWKYKEG 365
>SPOT_AQUAE (O67012) Guanosine-3',5'-bis(diphosphate)
3'-pyrophosphohydrolase (EC 3.1.7.2) ((ppGpp)ase)
(Penta-phosphate guanosine-3'-pyrophosphohydrolase)
Length = 696
Score = 154 bits (389), Expect = 6e-37
Identities = 109/324 (33%), Positives = 173/324 (52%), Gaps = 26/324 (8%)
Query: 120 LSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESLRVKNFASR 179
L++A+ LA+L MD E I A +L + LE + EI+ + G A L+ ++ +
Sbjct: 54 LNVALKLAELGMDHETIIAALLHDTLEDTDTTYEEIKERFGERVAKLVEGVTKIGKIKYK 113
Query: 180 VDILDDENAAALRKFCL-TYYDIRALILDLALKLDMMRHLGHLPRYQQQIISLQVMKIYA 238
E A RK L T D R ++L L+ +LD ++ L +++ I+ + M+IYA
Sbjct: 114 -----SEQAENYRKLILATAEDPRVILLKLSDRLDNVKTLWVFREEKRKKIAKETMEIYA 168
Query: 239 PLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKDELLESLKSD 298
PLAH +G I ELED +F+YL+P Y V +++ +L + + ++ ++ +
Sbjct: 169 PLAHRLGVWSIKNELEDWAFKYLYPEEYEKVRNFVKESRK---NLEEYLRKYVIPKVRKE 225
Query: 299 PILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSRENALEAGER 358
L + + +K R K YS +K + G R EDV+D+LG+R+++N
Sbjct: 226 --LEKYGIEAEIKYRSKHYYSIWEKTRRKGIRLEDVHDILGVRIIVNTVPE--------- 274
Query: 359 ACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLMEIQIRTTEM 418
CY II S+++ +P + KDYIS PK N Y+SLH V +++ G+ L+E QIRT EM
Sbjct: 275 -CYTVLGIIHSLFRPVPGKFKDYISLPKPNLYQSLHTTV-IADKGK---LVEFQIRTWEM 329
Query: 419 DRLAVGGMASHSLYKAGLTNPEEA 442
A G+ASH YK G NP +A
Sbjct: 330 HERAEKGIASHWAYKEG-KNPSDA 352
>SPOT_BORBU (O51216) Guanosine-3',5'-bis(diphosphate)
3'-pyrophosphohydrolase (EC 3.1.7.2) ((ppGpp)ase)
(Penta-phosphate guanosine-3'-pyrophosphohydrolase)
Length = 667
Score = 153 bits (386), Expect = 1e-36
Identities = 104/342 (30%), Positives = 176/342 (51%), Gaps = 21/342 (6%)
Query: 92 LFKSLKLSIPVLQTSPLTPDGRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELN 151
+FK+L+++ L G + + +++ LA Q+D + AG+L +VLE +
Sbjct: 40 IFKALEIA-EQLHYGQYRESGEPYIIHPIMVSLFLAKFQLDFKATIAGLLHDVLEDTNVE 98
Query: 152 LHEIRSQIGSATAHLLHESLRVKNFASRVDILDDENAAALRKFCLTYYDIRALILDLALK 211
EI + L+ ++ + ++ + + N + F +T+ DIR +I+ LA K
Sbjct: 99 KEEIVKEFDEEILSLIDGVTKIHDLHNKTRSIKEANTISKMFFAMTH-DIRIIIIKLADK 157
Query: 212 LDMMRHLGHLPRYQQQIISLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDT 271
L M L +LP+ +Q I+ + Y P+A +G + + LEDLSF++L+P Y +
Sbjct: 158 LHNMTTLSYLPKNRQDRIAKDCLSTYVPIAERLGISSLKTYLEDLSFKHLYPKDYKEIKN 217
Query: 272 WLRSQETGGISLIDVYKDELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRP 331
+L ET +YK +L S++ + + + +I+V R K YS +K+ +
Sbjct: 218 FL--SETKIEREKKLYKGKL--SIEKELQKSGIEAEITV--RSKHFYSIFRKMQTRTNKL 271
Query: 332 EDVNDVLGLRVVLNPKSRENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYR 391
+ D LG+R++ ++ CY +I+ +WK IP R KDYI+ PK N Y+
Sbjct: 272 TQIFDTLGIRIICKK----------QKECYEILEIVHRVWKPIPGRLKDYIASPKENKYQ 321
Query: 392 SLHMAVDVSEIGRTRPLMEIQIRTTEMDRLAVGGMASHSLYK 433
SLH V + E L+EIQIRT EMDR+A G+A+H +YK
Sbjct: 322 SLHTTVRIPE---DNQLIEIQIRTEEMDRIANYGVAAHWIYK 360
>SPOT_SYNY3 (P74007) Probable guanosine-3',5'-bis(diphosphate)
3'-pyrophosphohydrolase (EC 3.1.7.2) ((ppGpp)ase)
(Penta-phosphate guanosine-3'-pyrophosphohydrolase)
Length = 760
Score = 151 bits (382), Expect = 4e-36
Identities = 107/324 (33%), Positives = 168/324 (51%), Gaps = 24/324 (7%)
Query: 112 GRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESL 171
G ++ +++A LL DL D +I+AG L +V+E ++++ +I + G TA L+
Sbjct: 73 GEPYIAHPVAVAGLLRDLGGDEAMIAAGFLHDVVEDTDISIEQIEALFGEETASLVEGVT 132
Query: 172 RVK--NFASRVDILDDENAAALRKFCLTYYDIRALILDLALKLDMMRHLGHLPRYQQQII 229
++ NF+S + EN R F DIR +++ LA +L MR L L +Q+ I
Sbjct: 133 KLSKFNFSSTTEH-QAENFR--RMFLAMAKDIRVIVVKLADRLHNMRTLDALSPEKQRRI 189
Query: 230 SLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKD 289
+ + I+APLA+ +G ELEDLSF+YL P SY + + + + S ++ KD
Sbjct: 190 ARETKDIFAPLANRLGIWRFKWELEDLSFKYLEPDSYRKIQSLVVEKRGDRESRLETVKD 249
Query: 290 ELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSR 349
L L+ E ++ ++GR K Y K+ + E++ D+ LR+++ K
Sbjct: 250 MLRFRLRD-----EGIEHFELQGRPKHLYGIYYKMTSQDKAFEEIYDIAALRIIVESKGE 304
Query: 350 ENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLM 409
CYRA ++ ++K IP R KDYI PK N Y+SLH V +G T +
Sbjct: 305 ----------CYRALSVVHDVFKPIPGRFKDYIGLPKPNRYQSLHTTV----LGLTSRPL 350
Query: 410 EIQIRTTEMDRLAVGGMASHSLYK 433
EIQIRT EM +A G+A+H YK
Sbjct: 351 EIQIRTEEMHHVAEYGIAAHWKYK 374
>RELA_STRCO (P52560) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP
3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 847
Score = 148 bits (374), Expect = 3e-35
Identities = 102/319 (31%), Positives = 165/319 (50%), Gaps = 25/319 (7%)
Query: 116 LSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESLRVKN 175
++ L++ +LA+L MD + AG+L + +E E L ++R G L+ ++
Sbjct: 150 ITHPLAVTTILAELGMDPATLMAGLLHDTVEDTEYGLEDLRRDFGDVVTLLVDGVTKL-- 207
Query: 176 FASRVDILDDENAAALRKFCLTYY-DIRALILDLALKLDMMRHLGHLPRYQQQIISLQVM 234
+V + A +RK + D R L++ LA +L MR + +L R +Q+ + + +
Sbjct: 208 --DKVKFGEAAQAETVRKMVVAMAKDPRVLVIKLADRLHNMRTMRYLKREKQEKKARETL 265
Query: 235 KIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKDELLES 294
+IYAPLAH +G N I ELEDL+F L+P Y + + + + V DE+ +
Sbjct: 266 EIYAPLAHRLGMNTIKWELEDLAFAILYPKMYDEIVRLVAERAPKRDEYLAVVTDEVQQD 325
Query: 295 LKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSRENALE 354
L++ I A +V GR K YS +K++ GR ++ D++G+RV+++
Sbjct: 326 LRAARIKA------TVTGRPKHYYSVYQKMIVRGRDFAEIYDLVGIRVLVDT-------- 371
Query: 355 AGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLMEIQIR 414
R CY A + + W +P R KDYI+ PK N Y+SLH V IG +E+QIR
Sbjct: 372 --VRDCYAALGTVHARWNPVPGRFKDYIAMPKFNMYQSLHTTV----IGPGGKPVELQIR 425
Query: 415 TTEMDRLAVGGMASHSLYK 433
T +M R A G+A+H YK
Sbjct: 426 TFDMHRRAEYGIAAHWKYK 444
>RELA_STAAW (O32419) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP
3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 736
Score = 147 bits (371), Expect = 7e-35
Identities = 98/338 (28%), Positives = 173/338 (50%), Gaps = 23/338 (6%)
Query: 120 LSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESLRVKNFASR 179
+ +A +L ++++D I AG L +V+E +++ A ++ ++K R
Sbjct: 62 IQVAGILTEMRLDGPTIVAGFLHDVIEDTPYTFEDVKEMFNEEVARIVDGVTKLKKVKYR 121
Query: 180 VDILDDENAAALRK-FCLTYYDIRALILDLALKLDMMRHLGHLPRYQQQIISLQVMKIYA 238
+++ A RK F D+R +++ LA +L MR L +PR +Q IS + ++IYA
Sbjct: 122 SK--EEQQAENHRKLFIAIAKDVRVILVKLADRLHNMRTLKAMPREKQIRISRETLEIYA 179
Query: 239 PLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKDELLESLKSD 298
PLAH +G N I ELED + +Y+ Y + ++ + S + Y + ++ ++++
Sbjct: 180 PLAHRLGINTIKWELEDTALRYIDNVQYFRIVNLMKKKR----SEREAYIETAIDRIRTE 235
Query: 299 PILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSRENALEAGER 358
+ DI+ GR K YS +K++K ++ + + D+L +RV++N
Sbjct: 236 MDRMNIEGDIN--GRPKHIYSIYRKMMKQKKQFDQIFDLLAIRVIVN----------SIN 283
Query: 359 ACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLMEIQIRTTEM 418
CY ++ ++WK +P R KDYI+ PK N Y+SLH V +G +EIQIRT +M
Sbjct: 284 DCYAILGLVHTLWKPMPGRFKDYIAMPKQNLYQSLHTTV----VGPNGDPLEIQIRTFDM 339
Query: 419 DRLAVGGMASHSLYKAGLTNPEEAKRLKTIMLAAAELA 456
+A G+A+H YK G E+ + + + ELA
Sbjct: 340 HEIAEHGVAAHWAYKEGKKVSEKDQTYQNKLNWLKELA 377
>RELA_STREQ (Q54089) Putative GTP pyrophosphokinase (EC 2.7.6.5)
(ATP:GTP 3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase) (Stringent response-like protein)
Length = 739
Score = 146 bits (368), Expect = 2e-34
Identities = 106/341 (31%), Positives = 172/341 (50%), Gaps = 24/341 (7%)
Query: 120 LSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESLRVKNFASR 179
+ +A +LADL +DA ++ G L +V+E ++ L I G ++ ++ +
Sbjct: 55 IQVAGILADLHLDAVTVACGFLHDVVEDTDITLDNIEFDFGKDVRDIVDGVTKLGKVEYK 114
Query: 180 VDILDDENAAALRKFCLTYY-DIRALILDLALKLDMMRHLGHLPRYQQQIISLQVMKIYA 238
+++ A RK + DIR +++ LA +L MR L HL + +Q+ IS + M+IYA
Sbjct: 115 SH--EEQLAENHRKMLMAMSKDIRVILVKLADRLHNMRTLKHLRKDKQERISRETMEIYA 172
Query: 239 PLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKDELLESLKSD 298
PLAH +G + I ELEDL+F+YL + + + + +L+ D+++ +KS
Sbjct: 173 PLAHRLGISRIKWELEDLAFRYLNETEFYKISHMMNEKRREREALV----DDIVTKIKSY 228
Query: 299 PILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSRENALEAGER 358
L D V GR K YS +K+ +R + + D++ +R V+ +S A+
Sbjct: 229 TTEQGLFGD--VYGRPKHIYSIYRKMRDKKKRFDQIFDLIAIRCVMETQSDVYAMVG--- 283
Query: 359 ACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLMEIQIRTTEM 418
I +W+ +P R KDYI+ PK NGY+S+H V G P+ EIQIRT EM
Sbjct: 284 -------YIHELWRPMPGRFKDYIAAPKANGYQSIHTTV----YGPKGPI-EIQIRTKEM 331
Query: 419 DRLAVGGMASHSLYKAGLTNPEEAKRLKTIMLAAAELAALR 459
++A G+A+H YK G+ K M EL L+
Sbjct: 332 HQVAEYGVAAHWAYKKGVRGKVNQAEQKVGMNWIKELVELQ 372
>RELA_STRAT (O85709) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP
3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 841
Score = 145 bits (367), Expect = 2e-34
Identities = 101/319 (31%), Positives = 163/319 (50%), Gaps = 25/319 (7%)
Query: 116 LSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESLRVKN 175
++ L++ +LA+L MD + AG+L + E E L ++R G L+ ++
Sbjct: 147 ITHPLAVTTILAELGMDPATLMAGLLHDSREDTEYGLDDLRRDFGDVVGLLVDGVTKL-- 204
Query: 176 FASRVDILDDENAAALRKFCLTYY-DIRALILDLALKLDMMRHLGHLPRYQQQIISLQVM 234
+V + A +RK + D R L++ LA +L MR + +L R +Q+ + + +
Sbjct: 205 --DKVKFGEAAQAETVRKMVVAMAKDPRVLVIKLADRLHNMRTMRYLKREKQEKKARETL 262
Query: 235 KIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKDELLES 294
+IYAPLAH +G N I ELEDL+F L+P Y + + + + + DE+
Sbjct: 263 EIYAPLAHRLGMNTIKWELEDLAFAILYPKMYDEIVRLVAERAPKRDEYLAIVTDEVQSD 322
Query: 295 LKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSRENALE 354
L++ I A +V GR K YS +K++ GR ++ D++G+RV+++
Sbjct: 323 LRARRIKA------TVTGRPKHYYSVYQKIIVRGRDFAEIYDLVGIRVLVDT-------- 368
Query: 355 AGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLMEIQIR 414
R CY A + + W +P R KDYI+ PK N Y+SLH V IG +E+QIR
Sbjct: 369 --VRDCYAALGTVHARWNPVPGRFKDYIAMPKFNMYQSLHTTV----IGPNGKPVELQIR 422
Query: 415 TTEMDRLAVGGMASHSLYK 433
T +M R A G+A+H YK
Sbjct: 423 TFDMHRRAEYGIAAHWKYK 441
>SPOT_SPICI (O34098) Guanosine-3',5'-bis(diphosphate)
3'-pyrophosphohydrolase (EC 3.1.7.2) ((ppGpp)ase)
(Penta-phosphate guanosine-3'-pyrophosphohydrolase)
Length = 749
Score = 145 bits (365), Expect = 4e-34
Identities = 106/326 (32%), Positives = 173/326 (52%), Gaps = 27/326 (8%)
Query: 126 LADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESLRVKNFA--SRVDIL 183
LA +M + + AG+L +VLE E++ G A+L+ +V FA +R I
Sbjct: 60 LAQWRMGPKTLIAGLLHDVLEDTPATFEELQELFGIEIANLVEGVTKVSYFAKENRTQI- 118
Query: 184 DDENAAALRKFCLTYY-DIRALILDLALKLDMMRHLGHLPRYQQQIISLQVMKIYAPLAH 242
A LRK L+ DIR +I+ LA +L ++ +G+L +QQII+ + ++IY+ +AH
Sbjct: 119 ---KAQYLRKLYLSMAKDIRVIIVKLADRLHNLKTIGYLKPERQQIIARESLEIYSAIAH 175
Query: 243 AVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKDELLESLKSDPILA 302
+G + E+ED+SF+ + P Y + + L S + I+ +EL + L +
Sbjct: 176 RLGMKAVKQEIEDISFKIINPVQYNKIVSLLESSNKERENTINQKIEELKKIL-----IT 230
Query: 303 ELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSRENALEAGERACYR 362
E + V GR KS YS +K+ + G+ +D++D+L +R++ N CY+
Sbjct: 231 EKKMSVKVYGRSKSIYSIYRKMNQFGKNFDDIHDILAVRIITNSVD----------DCYK 280
Query: 363 AHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLMEIQIRTTEMDRLA 422
+ + + +R KDYI+ PK N Y+SLH + V++ G + E+QIRT EMD LA
Sbjct: 281 VLGFVHQHYTPLNNRFKDYIATPKHNLYQSLHTTI-VADDGL---IFEVQIRTEEMDELA 336
Query: 423 VGGMASHSLYKAGLTNPEEAKRLKTI 448
G+A+H YK G N + AK+ K I
Sbjct: 337 EQGVAAHWRYKEG-ENYDIAKKQKDI 361
>SPOT_ECOLI (P17580) Guanosine-3',5'-bis(diphosphate)
3'-pyrophosphohydrolase (EC 3.1.7.2) ((ppGpp)ase)
(Penta-phosphate guanosine-3'-pyrophosphohydrolase)
Length = 702
Score = 140 bits (354), Expect = 7e-33
Identities = 103/348 (29%), Positives = 172/348 (48%), Gaps = 28/348 (8%)
Query: 112 GRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESL 171
G ++ +++A +LA++++D E + A +L +V+E ++ G + A L+
Sbjct: 42 GEPYITHPVAVACILAEMKLDYETLMAALLHDVIEDTPATYQDMEQLFGKSVAELVEGVS 101
Query: 172 RVKNFASRVDILDDENAAA--LRKFCLTYY-DIRALILDLALKLDMMRHLGHLPRYQQQI 228
++ R D + A A RK + DIR +++ LA + MR LG L +++
Sbjct: 102 KLDKLKFR----DKKEAQAENFRKMIMAMVQDIRVILIKLADRTHNMRTLGSLRPDKRRR 157
Query: 229 ISLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYK 288
I+ + ++IY+PLAH +G ++I ELE+L F+ L+P Y + +++ +I
Sbjct: 158 IARETLEIYSPLAHRLGIHHIKTELEELGFEALYPNRYRVIKEVVKAARGNRKEMIQKIL 217
Query: 289 DELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKS 348
E+ L+ I V GR K YS K++ +R + D+ RV++N
Sbjct: 218 SEIEGRLQEAGI------PCRVSGREKHLYSIYCKMVLKEQRFHSIMDIYAFRVIVNDSD 271
Query: 349 RENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPL 408
CYR + S++K P R KDYI+ PK NGY+SLH S IG
Sbjct: 272 ----------TCYRVLGQMHSLYKPRPGRVKDYIAIPKANGYQSLH----TSMIGPHGVP 317
Query: 409 MEIQIRTTEMDRLAVGGMASHSLYKA-GLTNPEEAKRLKTIMLAAAEL 455
+E+QIRT +MD++A G+A+H YK G T+ R + M + EL
Sbjct: 318 VEVQIRTEDMDQMAEMGVAAHWAYKEHGETSTTAQIRAQRWMQSLLEL 365
>RELA_MYCTU (P66014) Probable GTP pyrophosphokinase (EC 2.7.6.5)
(ATP:GTP 3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 790
Score = 135 bits (340), Expect = 3e-31
Identities = 97/346 (28%), Positives = 170/346 (49%), Gaps = 30/346 (8%)
Query: 106 SPLTPDGRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAH 165
S L G ++ L++A +LA+L MD + A +L + +E L + + G H
Sbjct: 96 SQLRQSGDPYITHPLAVANILAELGMDTTTLVAALLHDTVEDTGYTLEALTEEFGEEVGH 155
Query: 166 LLHESLRVKNFASRVDILDDENAAALRKFCLTYY-DIRALILDLALKLDMMRHLGHLPRY 224
L+ ++ RV + +RK D R L++ +A +L MR + LP
Sbjct: 156 LVDGVTKL----DRVVLGSAAEGETIRKMITAMARDPRVLVIKVADRLHNMRTMRFLPPE 211
Query: 225 QQQIISLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLI 284
+Q + + +++ APLAH +G + ELEDLSF L P Y + + + + +
Sbjct: 212 KQARKARETLEVIAPLAHRLGMASVKWELEDLSFAILHPKKYEEIVRLVAGRAPSRDTYL 271
Query: 285 DVYKDELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVL 344
+ E++ +L + I A +V+GR K +S +K++ GR +D++D++G+R++
Sbjct: 272 AKVRAEIVNTLTASKIKA------TVEGRPKHYWSIYQKMIVKGRDFDDIHDLVGVRILC 325
Query: 345 NPKSRENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGR 404
+ R CY A ++ S+W+ + R KDYI++P+ Y+SLH V +G
Sbjct: 326 DE----------IRDCYAAVGVVHSLWQPMAGRFKDYIAQPRYGVYQSLHTTV----VGP 371
Query: 405 TRPLMEIQIRTTEMDRLAVGGMASHSLYKA-----GLTNPEEAKRL 445
+E+QIRT +M R A G+A+H YK G+ +P A +
Sbjct: 372 EGKPLEVQIRTRDMHRTAEYGIAAHWRYKEAKGRNGVLHPHAAAEI 417
>RELA_MYCBO (P66015) Probable GTP pyrophosphokinase (EC 2.7.6.5)
(ATP:GTP 3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 790
Score = 135 bits (340), Expect = 3e-31
Identities = 97/346 (28%), Positives = 170/346 (49%), Gaps = 30/346 (8%)
Query: 106 SPLTPDGRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAH 165
S L G ++ L++A +LA+L MD + A +L + +E L + + G H
Sbjct: 96 SQLRQSGDPYITHPLAVANILAELGMDTTTLVAALLHDTVEDTGYTLEALTEEFGEEVGH 155
Query: 166 LLHESLRVKNFASRVDILDDENAAALRKFCLTYY-DIRALILDLALKLDMMRHLGHLPRY 224
L+ ++ RV + +RK D R L++ +A +L MR + LP
Sbjct: 156 LVDGVTKL----DRVVLGSAAEGETIRKMITAMARDPRVLVIKVADRLHNMRTMRFLPPE 211
Query: 225 QQQIISLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLI 284
+Q + + +++ APLAH +G + ELEDLSF L P Y + + + + +
Sbjct: 212 KQARKARETLEVIAPLAHRLGMASVKWELEDLSFAILHPKKYEEIVRLVAGRAPSRDTYL 271
Query: 285 DVYKDELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVL 344
+ E++ +L + I A +V+GR K +S +K++ GR +D++D++G+R++
Sbjct: 272 AKVRAEIVNTLTASKIKA------TVEGRPKHYWSIYQKMIVKGRDFDDIHDLVGVRILC 325
Query: 345 NPKSRENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGR 404
+ R CY A ++ S+W+ + R KDYI++P+ Y+SLH V +G
Sbjct: 326 DE----------IRDCYAAVGVVHSLWQPMAGRFKDYIAQPRYGVYQSLHTTV----VGP 371
Query: 405 TRPLMEIQIRTTEMDRLAVGGMASHSLYKA-----GLTNPEEAKRL 445
+E+QIRT +M R A G+A+H YK G+ +P A +
Sbjct: 372 EGKPLEVQIRTRDMHRTAEYGIAAHWRYKEAKGRNGVLHPHAAAEI 417
>RELA_MYCLE (Q49640) Probable GTP pyrophosphokinase (EC 2.7.6.5)
(ATP:GTP 3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 787
Score = 135 bits (339), Expect = 4e-31
Identities = 97/346 (28%), Positives = 170/346 (49%), Gaps = 30/346 (8%)
Query: 106 SPLTPDGRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAH 165
S L G ++ L++A +LA+L MD + A +L + +E L + + G H
Sbjct: 93 SQLRRSGDPYITHPLAVANILAELGMDITTLVAALLHDTVEDTGYTLEALSEEFGDEVGH 152
Query: 166 LLHESLRVKNFASRVDILDDENAAALRKFCLTYY-DIRALILDLALKLDMMRHLGHLPRY 224
L+ ++ RV + +RK D R L++ +A +L MR + LP
Sbjct: 153 LVDGVTKL----DRVVLGSAAEGETIRKMITAMARDPRVLVIKVADRLHNMRTMRFLPPE 208
Query: 225 QQQIISLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLI 284
+Q + + +++ APLAH +G + ELEDLSF L P Y + + + + +
Sbjct: 209 KQARKARETLEVIAPLAHRLGMASVKWELEDLSFAILHPKKYEEIVRLVAGRAPSRDTYL 268
Query: 285 DVYKDELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVL 344
+ E++ +L + I A +V+GR K +S +K++ GR +D++D++G+R++
Sbjct: 269 AKVRAEIISTLGASKIKA------TVEGRPKHYWSIYQKMIVKGRDFDDIHDLVGIRILC 322
Query: 345 NPKSRENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGR 404
+ R CY A ++ S+W+ + R KDYI++P+ Y+SLH V +G
Sbjct: 323 DE----------IRDCYAAVGVVHSLWQPMAGRFKDYIAQPRYGVYQSLHTTV----VGP 368
Query: 405 TRPLMEIQIRTTEMDRLAVGGMASHSLYKA-----GLTNPEEAKRL 445
+E+QIRT +M R A G+A+H YK G+ +P A +
Sbjct: 369 EGKPLEVQIRTRDMHRTAEYGIAAHWRYKEAKGRNGVLHPHAAAEI 414
>RELA_ECOLI (P11585) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP
3'-pyrophosphotransferase) (ppGpp synthetase I)
((p)ppGpp synthetase)
Length = 744
Score = 128 bits (322), Expect = 4e-29
Identities = 90/338 (26%), Positives = 161/338 (47%), Gaps = 25/338 (7%)
Query: 103 LQTSPLTPDGRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSA 162
LQ + PD L + + + +L+ L MD + + A +L + + ++ +R +G +
Sbjct: 43 LQQTQGHPDASLLLWRGVEMVEILSTLSMDIDTLRAALLFPLADANVVSEDVLRESVGKS 102
Query: 163 TAHLLH---ESLRVKNF-ASRVDILDDENAAALRKFCLTYYD-IRALILDLALKLDMMRH 217
+L+H + ++ A+ D + E +R+ L D R +++ LA ++ +R
Sbjct: 103 VVNLIHGVRDMAAIRQLKATHTDSVSSEQVDNVRRMLLAMVDDFRCVVIKLAERIAHLRE 162
Query: 218 LGHLPRYQQQIISLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQE 277
+ P ++ + + + IYAPLA+ +G + ELED F+YL P Y + L +
Sbjct: 163 VKDAPEDERVLAAKECTNIYAPLANRLGIGQLKWELEDYCFRYLHPTEYKRIAKLLHERR 222
Query: 278 TGGISLIDVYKDELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDV 337
I+ + L +K++ + AE V GR K YS +K+ K +++ DV
Sbjct: 223 LDREHYIEEFVGHLRAEMKAEGVKAE------VYGRPKHIYSIWRKMQKKNLAFDELFDV 276
Query: 338 LGLRVVLNPKSRENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAV 397
+R+V + CY A I+ + ++ +P DY++ PK NGY+S+H V
Sbjct: 277 RAVRIVAERL----------QDCYAALGIVHTHYRHLPDEFDDYVANPKPNGYQSIHTVV 326
Query: 398 DVSEIGRTRPLMEIQIRTTEMDRLAVGGMASHSLYKAG 435
+G +EIQIRT +M A G+A+H YK G
Sbjct: 327 ----LGPGGKTVEIQIRTKQMHEDAELGVAAHWKYKEG 360
>SPOT_HAEIN (P43811) Guanosine-3',5'-bis(diphosphate)
3'-pyrophosphohydrolase (EC 3.1.7.2) ((ppGpp)ase)
(Penta-phosphate guanosine-3'-pyrophosphohydrolase)
Length = 677
Score = 128 bits (321), Expect = 5e-29
Identities = 91/336 (27%), Positives = 164/336 (48%), Gaps = 23/336 (6%)
Query: 112 GRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESL 171
G ++ +++A ++A L +D E + A +L +V+E +++ + G++ A ++
Sbjct: 15 GEPYITHPVAVASIIAQLHLDHEAVMAALLHDVIEDTPYTEEQLKEEFGASVAEIVDGVS 74
Query: 172 RVKNFASRVDILDDENAAALRKFCLTYY-DIRALILDLALKLDMMRHLGHLPRYQQQIIS 230
++ R + RK L DIR +++ LA + MR LG L +++ I+
Sbjct: 75 KLDKLKFRTR--QEAQVENFRKMILAMTRDIRVVLIKLADRTHNMRTLGSLRPDKRRRIA 132
Query: 231 LQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKDE 290
+ ++IY PLAH +G +I ELEDLSFQ + P+ Y + + + LI+ E
Sbjct: 133 KETLEIYCPLAHRLGIEHIKNELEDLSFQAMHPHRYEVLKKLVDVARSNRQDLIERISQE 192
Query: 291 LLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSRE 350
+ L++ I A V GR K Y +K+ + + D+ RV++ K+ +
Sbjct: 193 IKVRLENSGIFAR------VWGREKHLYKIYQKMRIKDQEFHSIMDIYAFRVIV--KNVD 244
Query: 351 NALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLME 410
+ CYR + +++K P R KDYI+ PK NGY+SL S IG +E
Sbjct: 245 D--------CYRVLGQMHNLYKPRPGRVKDYIAVPKANGYQSL----QTSMIGPKGVPVE 292
Query: 411 IQIRTTEMDRLAVGGMASHSLYKAGLTNPEEAKRLK 446
+ I T +M+++A G+ +H +YK N +++
Sbjct: 293 VHIHTEDMEQVAEMGITAHWVYKENGKNDSTTAQIR 328
>RELA_CORGL (O87331) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP
3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 760
Score = 125 bits (313), Expect = 4e-28
Identities = 91/332 (27%), Positives = 162/332 (48%), Gaps = 25/332 (7%)
Query: 103 LQTSPLTPDGRSPLSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSA 162
L + G ++ L++A + A++ MD + A +L + +E + +L ++ G
Sbjct: 65 LHDGVIRKSGDPYITHPLAVATIAAEIGMDTTTLVAALLHDTVEDTDYSLDDLTRDFGEE 124
Query: 163 TAHLLHESLRVKNFASRVDILDDENAAALRKFCLTY-YDIRALILDLALKLDMMRHLGHL 221
A L+ ++ +V + A +RK + D R L++ +A +L MR + L
Sbjct: 125 VARLVDGVTKL----DKVALGAAAEAETIRKMIVAMSQDPRVLVIKVADRLHNMRTMRFL 180
Query: 222 PRYQQQIISLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGI 281
P +Q + Q +++ APLAH +G + ELEDLSF L+P Y + + +
Sbjct: 181 PPEKQAKKARQTLEVIAPLAHRLGMASVKWELEDLSFAILYPKKYEEIVRLVADRAPSRD 240
Query: 282 SLIDVYKDELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLR 341
+ D++ L+ + I AE V GR K +S +K++ GR +D+ D++G+R
Sbjct: 241 RYLKEIIDQVTGGLRENNIAAE------VLGRPKHYWSIYQKMIVRGRDFDDIFDLVGIR 294
Query: 342 VVLNPKSRENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSE 401
++++ + CY A ++ S++ +P R KDYIS P+ Y+SLH V
Sbjct: 295 ILVDNVNN----------CYAAIGVVHSLFNALPGRFKDYISAPRFGVYQSLHTTV---- 340
Query: 402 IGRTRPLMEIQIRTTEMDRLAVGGMASHSLYK 433
+G +E+Q RT +M A G+A+H YK
Sbjct: 341 MGPGGKPLEVQARTHDMHYNAEFGIAAHWRYK 372
>RELA_VIBSS (P55133) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP
3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 744
Score = 119 bits (299), Expect = 2e-26
Identities = 86/326 (26%), Positives = 158/326 (48%), Gaps = 27/326 (8%)
Query: 116 LSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESLRVKN 175
L + + +L L MDA+ + A +L ++E G + ++ + HL+ + +
Sbjct: 55 LWRGRELVEILVTLSMDADTLIAALLYPLVEGGCYSTDALKEEYSGTILHLV-QGVEQMC 113
Query: 176 FASRVDILDDENAAA-----LRKFCLTYYD-IRALILDLALKLDMMRHLGHLPRYQQQII 229
S++ +E A A +R+ L+ D R +++ LA ++ +R + P ++
Sbjct: 114 AISQLKSTAEETAQAAQVDNIRRMLLSMVDDFRCVVIKLAERICNLREVKDQPDEVRRAA 173
Query: 230 SLQVMKIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKD 289
+ + IYAPLA+ +G + E+ED +F+Y P +Y + L + I + D
Sbjct: 174 AQECANIYAPLANRLGIGQLKWEIEDYAFRYQHPDTYKQIAKQLSERRIDREDYITHFVD 233
Query: 290 ELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSR 349
+L +++K+ I AE V+GR K YS +K+ K +++ DV +R+V
Sbjct: 234 DLSDAMKASNIRAE------VQGRPKHIYSIWRKMQKKSLEFDELFDVRAVRIVAEEL-- 285
Query: 350 ENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLM 409
+ CY A ++ + ++ +P DY++ PK NGY+S+H V +G +
Sbjct: 286 --------QDCYAALGVVHTKYRHLPKEFDDYVANPKPNGYQSIHTVV----LGPEGKTI 333
Query: 410 EIQIRTTEMDRLAVGGMASHSLYKAG 435
EIQIRT +M + G+A+H YK G
Sbjct: 334 EIQIRTKQMHEESELGVAAHWKYKEG 359
>RELA_HAEIN (P44644) GTP pyrophosphokinase (EC 2.7.6.5) (ATP:GTP
3'-pyrophosphotransferase) (ppGpp synthetase I)
((P)ppGpp synthetase)
Length = 743
Score = 118 bits (295), Expect = 5e-26
Identities = 91/341 (26%), Positives = 156/341 (45%), Gaps = 28/341 (8%)
Query: 116 LSKALSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLH-----ES 170
L + + +L +L MDAE + +L ++ + ++ + G+ LL ++
Sbjct: 59 LQSGVEMVEILHELNMDAETLLTAMLFPIVANKLTDWESLKEKFGAKITKLLKGVLEMDN 118
Query: 171 LRVKNFASRVDILDDENAAALRKFCLTYYDIRALILDLALKLDMMRHLGHLPRYQQQIIS 230
+R N + + L +N R D R +I+ LA ++ +R H + ++++
Sbjct: 119 IRQLNASHSANALQVDNVR--RMLLAMVDDFRCVIIKLAERITFLRDAEHRCAEEDKVLA 176
Query: 231 LQVMK-IYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKD 289
++ IYAPLA+ +G + ELED F+YL P Y + L+ + I +
Sbjct: 177 VKECSYIYAPLANRLGIGQLKWELEDYCFRYLHPEQYRAIAKLLQERRLDREHYIADFVS 236
Query: 290 ELLESLKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSR 349
EL L+ E ++ + V GR K YS +K+ K + DV +R+++
Sbjct: 237 ELSGYLR------ENIEQVEVYGRPKHIYSIWRKMQKKHLEFSGLYDVRAVRIIVQKL-- 288
Query: 350 ENALEAGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLM 409
+ CY A I+ + +K +P DY++ PK NGY+S+H V +G+ +
Sbjct: 289 --------QDCYTALGIVHTQFKHLPKEFDDYVANPKPNGYQSIHTVV----LGKGGKPI 336
Query: 410 EIQIRTTEMDRLAVGGMASHSLYKAGLTNPEEAKRLKTIML 450
E+QIRT +M A GMA+H YK G T A K L
Sbjct: 337 EVQIRTQQMHDDAELGMAAHWKYKEGNTGSMSAYEEKIAWL 377
>SPOT_MYCPN (P75386) Probable guanosine-3',5'-bis(diphosphate)
3'-pyrophosphohydrolase (EC 3.1.7.2) ((ppGpp)ase)
(Penta-phosphate guanosine-3'-pyrophosphohydrolase)
Length = 733
Score = 108 bits (269), Expect = 5e-23
Identities = 81/319 (25%), Positives = 157/319 (48%), Gaps = 26/319 (8%)
Query: 120 LSIAMLLADLQMDAEVISAGILREVLEVGELNLHEIRSQIGSATAHLLHESLRVKNFASR 179
L A+ L + MD+ + AG+L +++E ++ ++ + G L+ + ++ + + +
Sbjct: 67 LRTALRLVEWNMDSNTVCAGLLHDIIEDTQVTEADLTAIFGKEITDLVVKVTKITSESKK 126
Query: 180 VDIL----DDENAAALRKFCLT-YYDIRALILDLALKLDMMRHLGHLPRYQQQIISLQVM 234
L +D N +L ++ ++ AL+L LA +LD + + L +Q+II+ + +
Sbjct: 127 QRQLNRKKEDLNLKSLVNIAMSSQQEVNALVLKLADRLDNISSIEFLAVEKQKIIAKETL 186
Query: 235 KIYAPLAHAVGTNYISLELEDLSFQYLFPYSYLYVDTWLRSQETGGISLIDVYKDELLES 294
++YA +A +G + +L DLSF+ L P ++ + + Q+ + +K +L E
Sbjct: 187 ELYAKIAGRIGMYPVKTQLADLSFKVLDPKNFNNTLSKINQQKVFYDNEWGNFKKQLEEM 246
Query: 295 LKSDPILAELVDDISVKGRYKSRYSTMKKLLKDGRRPEDVNDVLGLRVVLNPKSRENALE 354
L+ + I + ++ R K YST +KL + ++D+ +R+++
Sbjct: 247 LEQNQI------EYRLESRIKGIYSTYQKLTFHEQNIAKIHDLFAIRLIVK--------- 291
Query: 355 AGERACYRAHQIIQSMWKEIPSRTKDYISRPKGNGYRSLHMAVDVSEIGRTRPLMEIQIR 414
E CY +I + + KDYI+ PK N Y+S+H V + + +EIQIR
Sbjct: 292 -SELDCYHLLGLIHLNFTVLMKHFKDYIASPKQNFYQSIHTTVRLKGLN-----VEIQIR 345
Query: 415 TTEMDRLAVGGMASHSLYK 433
T MD ++ G ASH +YK
Sbjct: 346 TQRMDHVSKYGFASHWIYK 364
Database: sprot
Posted date: Nov 25, 2004 10:54 AM
Number of letters in database: 59,974,054
Number of sequences in database: 164,201
Lambda K H
0.319 0.135 0.379
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 66,002,391
Number of Sequences: 164201
Number of extensions: 2770336
Number of successful extensions: 9824
Number of sequences better than 10.0: 368
Number of HSP's better than 10.0 without gapping: 196
Number of HSP's successfully gapped in prelim test: 174
Number of HSP's that attempted gapping in prelim test: 9056
Number of HSP's gapped (non-prelim): 749
length of query: 580
length of database: 59,974,054
effective HSP length: 116
effective length of query: 464
effective length of database: 40,926,738
effective search space: 18990006432
effective search space used: 18990006432
T: 11
A: 40
X1: 16 ( 7.4 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 41 (21.7 bits)
S2: 69 (31.2 bits)
Medicago: description of AC146862.24