
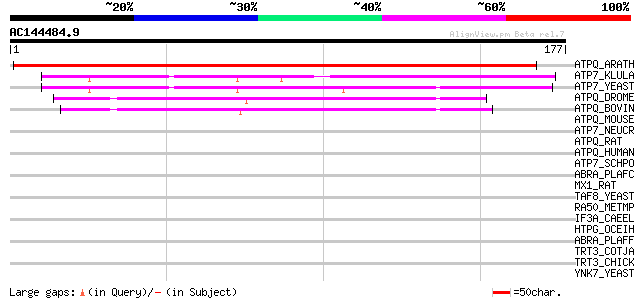
BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query= AC144484.9 + phase: 0
(177 letters)
Database: sprot
164,201 sequences; 59,974,054 total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
ATPQ_ARATH (Q9FT52) ATP synthase D chain, mitochondrial (EC 3.6.... 288 6e-78
ATP7_KLULA (O13350) ATP synthase D chain, mitochondrial (EC 3.6.... 57 2e-08
ATP7_YEAST (P30902) ATP synthase D chain, mitochondrial (EC 3.6.... 52 7e-07
ATPQ_DROME (Q24251) ATP synthase D chain, mitochondrial (EC 3.6.... 47 3e-05
ATPQ_BOVIN (P13620) ATP synthase D chain, mitochondrial (EC 3.6.... 42 7e-04
ATPQ_MOUSE (Q9DCX2) ATP synthase D chain, mitochondrial (EC 3.6.... 42 0.001
ATP7_NEUCR (Q7SI16) ATP synthase D chain, mitochondrial (EC 3.6.... 42 0.001
ATPQ_RAT (P31399) ATP synthase D chain, mitochondrial (EC 3.6.3.14) 41 0.001
ATPQ_HUMAN (O75947) ATP synthase D chain, mitochondrial (EC 3.6.... 40 0.002
ATP7_SCHPO (O94390) ATP synthase D chain, mitochondrial (EC 3.6.... 38 0.014
ABRA_PLAFC (P22620) 101 kDa malaria antigen (P101) (Acidic basic... 37 0.031
MX1_RAT (P18588) Interferon-induced GTP-binding protein Mx1 36 0.041
TAF8_YEAST (Q03750) Transcription initiation factor TFIID subuni... 36 0.053
RA50_METMP (P62134) DNA double-strand break repair rad50 ATPase 35 0.069
IF3A_CAEEL (P34339) Probable eukaryotic translation initiation f... 35 0.090
HTPG_OCEIH (Q8CX68) Chaperone protein htpG (Heat shock protein h... 35 0.090
ABRA_PLAFF (P23746) 101 kDa malaria antigen (P101) (Acidic basic... 35 0.12
TRT3_COTJA (P06398) Troponin T, fast skeletal muscle isoforms 34 0.20
TRT3_CHICK (P12620) Troponin T, fast skeletal muscle isoforms 34 0.20
YNK7_YEAST (P53930) Hypothetical 26.0 kDa protein in CYB5-LEU4 i... 33 0.26
>ATPQ_ARATH (Q9FT52) ATP synthase D chain, mitochondrial (EC
3.6.3.14)
Length = 167
Score = 288 bits (736), Expect = 6e-78
Identities = 136/167 (81%), Positives = 152/167 (90%)
Query: 2 SGTVKKVTDVAFKAGKKIDWDGMAKLLVTDEARREFFNLRRAFDEVNTQLETKFSQEPEP 61
SG KK+ DVAFKA + IDWDGMAK+LVTDEARREF NLRRAFDEVNTQL+TKFSQEPEP
Sbjct: 1 SGAGKKIADVAFKASRTIDWDGMAKVLVTDEARREFSNLRRAFDEVNTQLQTKFSQEPEP 60
Query: 62 IDWEYYRKGIGTRLVDMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKES 121
IDW+YYRKGIG +VD YK+ Y+SIEIPK+VD VTP+YKPKFDALLVELKEAE+KSLKES
Sbjct: 61 IDWDYYRKGIGAGIVDKYKEAYDSIEIPKYVDKVTPEYKPKFDALLVELKEAEQKSLKES 120
Query: 122 ERLEKEIVNVQSLKKRISTMTADEYFAEHPELKKKFDDEIRNDNWGY 168
ERLEKEI +VQ + K++STMTADEYF +HPELKKKFDDEIRNDNWGY
Sbjct: 121 ERLEKEIADVQEISKKLSTMTADEYFEKHPELKKKFDDEIRNDNWGY 167
>ATP7_KLULA (O13350) ATP synthase D chain, mitochondrial (EC
3.6.3.14)
Length = 173
Score = 57.4 bits (137), Expect = 2e-08
Identities = 40/171 (23%), Positives = 79/171 (45%), Gaps = 13/171 (7%)
Query: 11 VAFKAGKKIDWDGM-AKLLVTDEARREFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRK 69
+A A K+DW + + L +T + + + ++ DE QL + +P +D+ +YR
Sbjct: 2 LAKSAANKLDWAKVISSLKLTGKTATQLSSFKKRNDEARRQL-LELQSQPTSVDFSHYRS 60
Query: 70 GI-GTRLVDMYKQHYES-----IEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESER 123
+ T +VD +Q Y+S +++ K + T+ F++ +E EK + + +
Sbjct: 61 VLKNTEVVDKIEQFYKSYKPVSVDVSKQLSTIEA-----FESQAIENAAETEKLVAQELK 115
Query: 124 LEKEIVNVQSLKKRISTMTADEYFAEHPELKKKFDDEIRNDNWGYSHYKEE 174
KE +N + +T DE PE+ K ++ ++ W YKE+
Sbjct: 116 DLKETLNNIESARPFDQLTVDELTKARPEIDAKVEEMVKKGRWDVPGYKEK 166
>ATP7_YEAST (P30902) ATP synthase D chain, mitochondrial (EC
3.6.3.14)
Length = 173
Score = 52.0 bits (123), Expect = 7e-07
Identities = 37/166 (22%), Positives = 74/166 (44%), Gaps = 5/166 (3%)
Query: 11 VAFKAGKKIDWDGM-AKLLVTDEARREFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRK 69
+A A K+DW + + L +T + + ++ DE QL + +P +D+ +YR
Sbjct: 2 LAKSAANKLDWAKVISSLRITGSTATQLSSFKKRNDEARRQL-LELQSQPTEVDFSHYRS 60
Query: 70 GI-GTRLVDMYKQHYESIEIPKFVDTVTPQYKPKFDA-LLVELKEAEEKSLKESERLEKE 127
+ T ++D + + + + K + Q F+ + KE E KE + L+
Sbjct: 61 VLKNTSVIDKIESYVKQYKPVKIDASKQLQVIESFEKHAMTNAKETESLVSKELKDLQST 120
Query: 128 IVNVQSLKKRISTMTADEYFAEHPELKKKFDDEIRNDNWGYSHYKE 173
+ N+QS + +T D+ PE+ K ++ ++ W YK+
Sbjct: 121 LDNIQSARP-FDELTVDDLTKIKPEIDAKVEEMVKKGKWDVPGYKD 165
>ATPQ_DROME (Q24251) ATP synthase D chain, mitochondrial (EC
3.6.3.14)
Length = 178
Score = 46.6 bits (109), Expect = 3e-05
Identities = 32/139 (23%), Positives = 67/139 (48%), Gaps = 4/139 (2%)
Query: 15 AGKKIDWDGMAKLLVTDEARREFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRKGIGTR 74
A I+W +A+ + ++ + F + ++ + + P IDW Y+K +
Sbjct: 7 AQSSINWSALAERVPANQ--KSSFGAFKTKSDIYVRAVLANPECPPQIDWANYKKLVPVA 64
Query: 75 -LVDMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESERLEKEIVNVQS 133
LVD +++ YE++++P D V+ Q + A E+ ++ S + + +KEI +++S
Sbjct: 65 GLVDSFQKQYEALKVPYPQDKVSSQVDAEIKASQSEIDAYKKASEQRIQNYQKEIAHLKS 124
Query: 134 LKKRISTMTADEYFAEHPE 152
L MT ++Y P+
Sbjct: 125 LLP-YDQMTMEDYRDAFPD 142
>ATPQ_BOVIN (P13620) ATP synthase D chain, mitochondrial (EC
3.6.3.14)
Length = 160
Score = 42.0 bits (97), Expect = 7e-04
Identities = 30/139 (21%), Positives = 63/139 (44%), Gaps = 4/139 (2%)
Query: 17 KKIDWDGMAKLLVTDEARREFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRKGIG-TRL 75
K IDW +++ ++ + N ++++E T ++P IDW YY+ + L
Sbjct: 8 KTIDWVAFGEIIPRNQ--KAVANSLKSWNETLTSRLATLPEKPPAIDWAYYKANVAKAGL 65
Query: 76 VDMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESERLEKEIVNVQSLK 135
VD +++ + ++++P D T Q + + E +S + EKE+ ++++
Sbjct: 66 VDDFEKKFNALKVPIPEDKYTAQVDAEEKEDVKSCAEFLTQSKTRIQEYEKELEKMRNII 125
Query: 136 KRISTMTADEYFAEHPELK 154
MT ++ PE K
Sbjct: 126 P-FDQMTIEDLNEVFPETK 143
>ATPQ_MOUSE (Q9DCX2) ATP synthase D chain, mitochondrial (EC
3.6.3.14)
Length = 160
Score = 41.6 bits (96), Expect = 0.001
Identities = 35/147 (23%), Positives = 68/147 (45%), Gaps = 20/147 (13%)
Query: 17 KKIDWDGMAKLLVTDEARREFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRKGIG-TRL 75
K IDW +++ ++ + N ++++E S++P IDW YYR + L
Sbjct: 8 KTIDWVSFVEVMPQNQ--KAIGNALKSWNETFHARLASLSEKPPAIDWAYYRANVAKPGL 65
Query: 76 VDMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKE--------SERLEKE 127
VD +++ Y +++IP D K+ AL+ + ++ + KS E + EK+
Sbjct: 66 VDDFEKKYNALKIPVPED--------KYTALVDQEEKEDVKSCAEFVSGSQLRIQEYEKQ 117
Query: 128 IVNVQSLKKRISTMTADEYFAEHPELK 154
+ ++++ MT D+ PE K
Sbjct: 118 LEKMRNIIP-FDQMTIDDLNEIFPETK 143
>ATP7_NEUCR (Q7SI16) ATP synthase D chain, mitochondrial (EC
3.6.3.14)
Length = 173
Score = 41.6 bits (96), Expect = 0.001
Identities = 46/170 (27%), Positives = 75/170 (44%), Gaps = 15/170 (8%)
Query: 12 AFKAGKKIDWDGMAKLLVTDEARREFFNLRRAFDEVNTQLETKF---SQEPEPIDWEYYR 68
A A K+DW AK+ + R + +AF + N K S+ P +D+ YR
Sbjct: 3 ARNAALKVDW---AKITTSLGLRGQTAASLQAFKKRNDDARRKLQQLSELPTTVDFAAYR 59
Query: 69 KGIGTR-LVDMYKQHYESIEIPKFVDTVTPQYKPKFDALLVE-LKEAEEKSLK---ESER 123
+ + +V+ ++ + S + P D V Q K +A VE +K AE K E +
Sbjct: 60 STLKNQAIVNEIEKRFTSFK-PATYD-VNRQLKA-IEAFEVEAIKNAEATKTKVDLELKD 116
Query: 124 LEKEIVNVQSLKKRISTMTADEYFAEHPELKKKFDDEIRNDNWGYSHYKE 173
LEK + N+++ + +T DE A P + +K + W YKE
Sbjct: 117 LEKTLTNIETARP-FDELTVDEVAAAEPSIDEKTSKLVSKGRWSVPGYKE 165
>ATPQ_RAT (P31399) ATP synthase D chain, mitochondrial (EC 3.6.3.14)
Length = 160
Score = 41.2 bits (95), Expect = 0.001
Identities = 32/139 (23%), Positives = 59/139 (42%), Gaps = 4/139 (2%)
Query: 17 KKIDWDGMAKLLVTDEARREFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRKGIG-TRL 75
K IDW +++ ++ + N ++++E S++P IDW YYR + L
Sbjct: 8 KTIDWVSFVEIMPQNQ--KAIGNALKSWNETFHTRLASLSEKPPAIDWAYYRANVDKPGL 65
Query: 76 VDMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESERLEKEIVNVQSLK 135
VD +K Y +++IP D T + + + S EK++ ++++
Sbjct: 66 VDDFKNKYNALKIPVPEDKYTALVDAEEKEDVKNCAQFVTGSQARVREYEKQLEKIKNMI 125
Query: 136 KRISTMTADEYFAEHPELK 154
MT D+ PE K
Sbjct: 126 P-FDQMTIDDLNEVFPETK 143
>ATPQ_HUMAN (O75947) ATP synthase D chain, mitochondrial (EC
3.6.3.14) (My032 protein)
Length = 160
Score = 40.4 bits (93), Expect = 0.002
Identities = 31/139 (22%), Positives = 61/139 (43%), Gaps = 4/139 (2%)
Query: 17 KKIDWDGMAKLLVTDEARREFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRKGIG-TRL 75
K IDW A+++ ++ + + ++++E T + P IDW YY+ + L
Sbjct: 8 KTIDWVAFAEIIPQNQ--KAIASSLKSWNETLTSRLAALPENPPAIDWAYYKANVAKAGL 65
Query: 76 VDMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESERLEKEIVNVQSLK 135
VD +++ + ++++P D T Q + + E S EKE+ +++L
Sbjct: 66 VDDFEKKFNALKVPVPEDKYTAQVDAEEKEDVKSCAEWVSLSKARIVEYEKEMEKMKNLI 125
Query: 136 KRISTMTADEYFAEHPELK 154
MT ++ PE K
Sbjct: 126 P-FDQMTIEDLNEAFPETK 143
>ATP7_SCHPO (O94390) ATP synthase D chain, mitochondrial (EC
3.6.3.14)
Length = 174
Score = 37.7 bits (86), Expect = 0.014
Identities = 41/178 (23%), Positives = 71/178 (39%), Gaps = 27/178 (15%)
Query: 11 VAFKAGKKIDWDGMAKLLVTDEARRE-FFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRK 69
VA AGK IDW +A L D A N R + +L T ++ +D+ YR
Sbjct: 3 VASAAGKAIDWASVASKLKLDAATASAIANFRSRHAQAVAKLGT-LREQATTVDFATYRS 61
Query: 70 GIGTRLVDMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELK-------EAEEKSLKESE 122
+ + EI +++ +KP L +LK +A E + K E
Sbjct: 62 VLANK------------EIVNRIESSMKSFKPVKIDLNSQLKAINAFEAKASEGAKKNVE 109
Query: 123 RLEKEIVNVQSLKKRI------STMTADEYFAEHPELKKKFDDEIRNDNWGYSHYKEE 174
++ E+ N+ + K I +T ++ PE++K + + W Y+E+
Sbjct: 110 LVKAELQNLSATLKNIEQARPTEEITIEDMKQAVPEIEKIVETMVTKGKWVIPGYREK 167
>ABRA_PLAFC (P22620) 101 kDa malaria antigen (P101) (Acidic basic
repeat antigen)
Length = 743
Score = 36.6 bits (83), Expect = 0.031
Identities = 28/85 (32%), Positives = 43/85 (49%), Gaps = 2/85 (2%)
Query: 77 DMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSL-KESERLEKEIVNVQSLK 135
D+ Q E E+ K V+ +T Q + + DAL + KE EEK KE E+ EKE + +
Sbjct: 641 DVLNQETEE-EMEKQVEAITKQIEAEVDALAPKNKEEEEKEKEKEKEKEEKEKEEKEKEE 699
Query: 136 KRISTMTADEYFAEHPELKKKFDDE 160
K ++ E E KK+ ++E
Sbjct: 700 KEKEKEEKEKEKEEKEEEKKEKEEE 724
>MX1_RAT (P18588) Interferon-induced GTP-binding protein Mx1
Length = 652
Score = 36.2 bits (82), Expect = 0.041
Identities = 32/132 (24%), Positives = 56/132 (42%), Gaps = 24/132 (18%)
Query: 32 EARREFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRKGIGTR----LVDMYKQHYESIE 87
E R F LR F N +E E+++K +G+ ++ ++ HY E
Sbjct: 390 EESRLFTKLRNEFLAWNDYIE------------EHFKKTLGSSEKHSQMEKFESHYRGRE 437
Query: 88 IPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESERLEKEIVNVQSLKKRISTMTADEYF 147
+P FVD YK + + E+K EE +L R+ + N + ++S+ ++
Sbjct: 438 LPGFVD-----YKAFENIIKKEVKALEEPALNMLHRVTTMVKNAFT---KVSSNNFGDFL 489
Query: 148 AEHPELKKKFDD 159
H K K +D
Sbjct: 490 NLHSTAKSKIED 501
>TAF8_YEAST (Q03750) Transcription initiation factor TFIID subunit 8
(TBP-associated factor 8) (TBP-associated factor 65 kDa)
(TAFII-65)
Length = 510
Score = 35.8 bits (81), Expect = 0.053
Identities = 33/152 (21%), Positives = 62/152 (40%), Gaps = 27/152 (17%)
Query: 17 KKIDWDGMAKLLVTDEARR-EFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRKGIGTRL 75
K + W +A L +E E N+ +E+N L E + W
Sbjct: 130 KLMSWSSLAALPHNEEDEEDELNNIEEQQNEINVLLPPSNPLEKQIPSW----------- 178
Query: 76 VDMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESERLEKEIVNVQSLK 135
+P F T ++ P+F+ + +LK +++ +KES+ EK ++N L
Sbjct: 179 ------------LPNFPPDHTYKFTPEFNHPITDLKTIKKEIVKESQESEKALLN---LN 223
Query: 136 KRISTMTADEYFAEHPELKKKFDDEIRNDNWG 167
K +S +++ + P L + E + + WG
Sbjct: 224 KSLSHISSASNTPQPPGLDDEDAIEQQLEIWG 255
>RA50_METMP (P62134) DNA double-strand break repair rad50 ATPase
Length = 993
Score = 35.4 bits (80), Expect = 0.069
Identities = 28/89 (31%), Positives = 43/89 (47%), Gaps = 11/89 (12%)
Query: 94 TVTPQYKPKFDALLVELKEAE---EKSLKESERLEKEIVNVQSLKKRISTMTADEYFAEH 150
T P+ + L E+ E+E E+ LK+ E LEK L+K + +E FAE+
Sbjct: 186 TQEPEILENLEKLKNEVSESEILKEEILKKYENLEK-----LKLEKNSEILQMEEKFAEN 240
Query: 151 PELKKKFDD---EIRNDNWGYSHYKEEQN 176
+LK+ D EI+N N ++K N
Sbjct: 241 NQLKENLKDIISEIKNINLEIQNFKNSLN 269
>IF3A_CAEEL (P34339) Probable eukaryotic translation initiation
factor 3 subunit 10 (eIF-3 theta) (eIF3a)
Length = 1076
Score = 35.0 bits (79), Expect = 0.090
Identities = 28/114 (24%), Positives = 55/114 (47%), Gaps = 11/114 (9%)
Query: 23 GMAKLLVTDEARREFFNLRRAFDEVNTQLETKFSQEPEPIDWEYYRKGIGTRLVDMYKQH 82
G+ + L ++ R+E ++ Q+ + P I+ + RK ++++ YK++
Sbjct: 543 GLVEGLDAEKRRKEILK------KIEGQVTSYEKNRPTEIERIHRRK----KMLENYKEN 592
Query: 83 YESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESERLEKEIVNVQSLKK 136
+E ++ K T Q K + A E+K +E++ KESER K+ + KK
Sbjct: 593 WERVKAEKTAAAATEQAKREEAARAEEMKRLDEQN-KESERKRKQAEQDEIQKK 645
>HTPG_OCEIH (Q8CX68) Chaperone protein htpG (Heat shock protein
htpG) (High temperature protein G)
Length = 625
Score = 35.0 bits (79), Expect = 0.090
Identities = 29/109 (26%), Positives = 50/109 (45%), Gaps = 15/109 (13%)
Query: 68 RKGIGTRLVDMYKQHYESIEIPKFVDTVTPQ-----------YKPKFDALLVELKEAEEK 116
++ IGT + K++ E +F+DT T Q Y K D +LKE E
Sbjct: 162 KQDIGTTITLYIKENQEEENYDEFLDTFTLQQIIKKYSDFIRYPIKMDVTESKLKEGSED 221
Query: 117 SLKESERLEKEIVN--VQSLKKRISTMTADEYFAEHPELKKKFDDEIRN 163
++ +E++ +N V KK S +T D+Y + E + FD +++
Sbjct: 222 EYEDV--VEEQTINTMVPIWKKNKSELTDDDYTNFYQEKRYGFDKPLKH 268
>ABRA_PLAFF (P23746) 101 kDa malaria antigen (P101) (Acidic basic
repeat antigen) (Fragment)
Length = 321
Score = 34.7 bits (78), Expect = 0.12
Identities = 27/81 (33%), Positives = 40/81 (49%), Gaps = 2/81 (2%)
Query: 77 DMYKQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSL-KESERLEKEIVNVQSLK 135
D+ Q E E+ K V+ +T Q + + DAL + KE EEK KE E+ EKE + K
Sbjct: 242 DVLNQETEE-EMEKQVEAITKQIEAEVDALAPKNKEEEEKEKEKEKEKEEKEKEEKEKEK 300
Query: 136 KRISTMTADEYFAEHPELKKK 156
+ ++ E E +KK
Sbjct: 301 EEKEKEEKEKEEKEEKEEEKK 321
>TRT3_COTJA (P06398) Troponin T, fast skeletal muscle isoforms
Length = 252
Score = 33.9 bits (76), Expect = 0.20
Identities = 21/83 (25%), Positives = 47/83 (56%), Gaps = 3/83 (3%)
Query: 80 KQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESERLEKEIVNVQSLKKRIS 139
+Q+ + IE+ +D+ + + K + LV LKE EK + +ER E++ + + K+R +
Sbjct: 59 RQNKDLIELQALIDSHF-EARRKEEEELVALKERIEK--RRAERAEQQRIRAEKEKERQA 115
Query: 140 TMTADEYFAEHPELKKKFDDEIR 162
+ ++ E + K+K +D+++
Sbjct: 116 RLAEEKARREEEDAKRKAEDDLK 138
>TRT3_CHICK (P12620) Troponin T, fast skeletal muscle isoforms
Length = 262
Score = 33.9 bits (76), Expect = 0.20
Identities = 21/83 (25%), Positives = 47/83 (56%), Gaps = 3/83 (3%)
Query: 80 KQHYESIEIPKFVDTVTPQYKPKFDALLVELKEAEEKSLKESERLEKEIVNVQSLKKRIS 139
+Q+ + IE+ +D+ + + K + LV LKE EK + +ER E++ + + K+R +
Sbjct: 69 RQNKDLIELQALIDSHF-EARRKEEEELVALKERIEK--RRAERAEQQRIRAEKEKERQA 125
Query: 140 TMTADEYFAEHPELKKKFDDEIR 162
+ ++ E + K+K +D+++
Sbjct: 126 RLAEEKARREEEDAKRKAEDDLK 148
>YNK7_YEAST (P53930) Hypothetical 26.0 kDa protein in CYB5-LEU4
intergenic region
Length = 226
Score = 33.5 bits (75), Expect = 0.26
Identities = 25/77 (32%), Positives = 36/77 (46%)
Query: 48 NTQLETKFSQEPEPIDWEYYRKGIGTRLVDMYKQHYESIEIPKFVDTVTPQYKPKFDALL 107
+T L T F + P+ D Y+ K + +L D Y SIE P F T T + + +
Sbjct: 39 HTHLWTIFVRGPQNEDISYFIKKVVFKLHDTYPNPVRSIEAPPFELTETGWGEFDINIKV 98
Query: 108 VELKEAEEKSLKESERL 124
++EA EK L RL
Sbjct: 99 YFVEEANEKVLNFYHRL 115
Database: sprot
Posted date: Nov 25, 2004 10:54 AM
Number of letters in database: 59,974,054
Number of sequences in database: 164,201
Lambda K H
0.313 0.132 0.375
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 20,859,017
Number of Sequences: 164201
Number of extensions: 867203
Number of successful extensions: 3492
Number of sequences better than 10.0: 107
Number of HSP's better than 10.0 without gapping: 26
Number of HSP's successfully gapped in prelim test: 81
Number of HSP's that attempted gapping in prelim test: 3420
Number of HSP's gapped (non-prelim): 143
length of query: 177
length of database: 59,974,054
effective HSP length: 103
effective length of query: 74
effective length of database: 43,061,351
effective search space: 3186539974
effective search space used: 3186539974
T: 11
A: 40
X1: 16 ( 7.2 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 42 (21.9 bits)
S2: 62 (28.5 bits)
Medicago: description of AC144484.9