
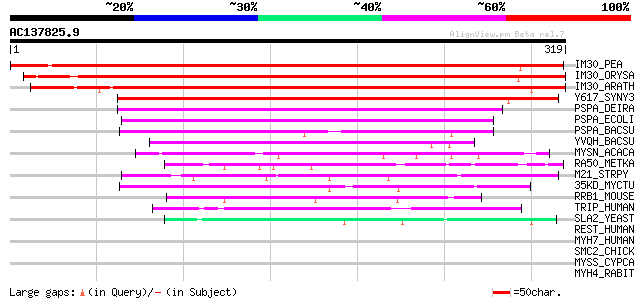
BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query= AC137825.9 - phase: 0
(319 letters)
Database: sprot
164,201 sequences; 59,974,054 total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
IM30_PEA (Q03943) Membrane-associated 30 kDa protein, chloroplas... 530 e-150
IM30_ORYSA (Q8S0J7) Probable membrane-associated 30 kDa protein,... 413 e-115
IM30_ARATH (O80796) Probable membrane-associated 30 kDa protein,... 411 e-114
Y617_SYNY3 (Q55707) Hypothetical protein sll0617 211 3e-54
PSPA_DEIRA (Q9RUB7) Phage shock protein A homolog 135 1e-31
PSPA_ECOLI (P23853) Phage shock protein A 116 8e-26
PSPA_BACSU (P54617) Phage shock protein A homolog 108 2e-23
YVQH_BACSU (O32201) Hypothetical protein yvqH 87 6e-17
MYSN_ACACA (P05659) Myosin II heavy chain, non muscle 50 7e-06
RA50_METKA (Q8TXI4) DNA double-strand break repair rad50 ATPase 50 1e-05
M21_STRPY (P50468) M protein, serotype 2.1 precursor 48 4e-05
35KD_MYCTU (P31511) 35 kDa protein 47 5e-05
RRB1_MOUSE (Q99PL5) Ribosome-binding protein 1 (Ribosome recepto... 47 6e-05
TRIP_HUMAN (Q9BWF2) TRAF-interacting protein 45 2e-04
SLA2_YEAST (P33338) SLA2 protein (Transmembrane protein MOP2) 44 4e-04
REST_HUMAN (P30622) Restin (Cytoplasmic linker protein-170 alpha... 43 0.001
MYH7_HUMAN (P12883) Myosin heavy chain, cardiac muscle beta isof... 43 0.001
SMC2_CHICK (Q90988) Structural maintenance of chromosome 2 (Chro... 43 0.001
MYSS_CYPCA (Q90339) Myosin heavy chain, fast skeletal muscle 43 0.001
MYH4_RABIT (Q28641) Myosin heavy chain, skeletal muscle, juvenile 43 0.001
>IM30_PEA (Q03943) Membrane-associated 30 kDa protein, chloroplast
precursor (M30)
Length = 323
Score = 530 bits (1366), Expect = e-150
Identities = 283/324 (87%), Positives = 300/324 (92%), Gaps = 8/324 (2%)
Query: 1 MATKLQIFTQLPSAPLQSSSSSSSLRKPLATSFFGTRPANTVKFRGMRIAKPVRGGGAIG 60
M TK QIF+ LPSAPLQ SSS L+KPLAT+ FGTRP +T+KFR MRIAKPVRGGGAIG
Sbjct: 1 MTTKFQIFSGLPSAPLQPSSSL--LKKPLATTLFGTRPVDTLKFRVMRIAKPVRGGGAIG 58
Query: 61 AGMNLFERFTRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRME 120
MNLF+RF RVVKSYANA+VS+FEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKR+E
Sbjct: 59 VRMNLFDRFARVVKSYANALVSTFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRLE 118
Query: 121 NKYKAATQASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSL 180
NKYKAA QASEEWYRKAQLALQKGEEDLAREALKRRKSFADNAS+LKAQLDQQK VVD+L
Sbjct: 119 NKYKAAQQASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASSLKAQLDQQKSVVDNL 178
Query: 181 VSNTRLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTM 240
VSNTRLLESKIQEARSKKDTLKARAQSAKT+TKVSEMLGNVNTS ALSAFEKMEEKVMTM
Sbjct: 179 VSNTRLLESKIQEARSKKDTLKARAQSAKTATKVSEMLGNVNTSSALSAFEKMEEKVMTM 238
Query: 241 ESQAEALGQLTSDDLEGKFAMLESSSVDDDLANLKKELSGSSKKGELPPGRS------ST 294
ESQAEALGQLT+DDLEGKFAMLE+SSVDDDLANLKKEL+GSSKKGELPPGRS ST
Sbjct: 239 ESQAEALGQLTTDDLEGKFAMLETSSVDDDLANLKKELAGSSKKGELPPGRSSTTSTTST 298
Query: 295 RTGTPFRDADIETELEQLRQRSKE 318
+TG PFRDADIE ELEQLR+RSKE
Sbjct: 299 KTGNPFRDADIEIELEQLRKRSKE 322
>IM30_ORYSA (Q8S0J7) Probable membrane-associated 30 kDa protein,
chloroplast precursor
Length = 317
Score = 413 bits (1062), Expect = e-115
Identities = 226/315 (71%), Positives = 262/315 (82%), Gaps = 9/315 (2%)
Query: 9 TQLPSAPLQSSSSSSSLRKPLATSFFGTRPANTVKFRGMRIAKP-VRGGGAIGAGMNLFE 67
T L AP +S+S R L TSF +V R +++ + V G NL +
Sbjct: 8 TSLRLAP-PPPASASFRRTALRTSFLN----GSVSLRLIQVRQSNVNRFKCNGIRSNLLD 62
Query: 68 RFTRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAAT 127
RF+RVVKSYANA++SSFEDPEKIL+QAVLEMNDDLTKMRQATAQVLASQKR+ENKYKAA
Sbjct: 63 RFSRVVKSYANAVLSSFEDPEKILDQAVLEMNDDLTKMRQATAQVLASQKRLENKYKAAE 122
Query: 128 QASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVSNTRLL 187
QAS++WYR+AQLALQKG+EDLAREALKRRKS+ADNAS+LKAQLDQQKGVV++LVSNTR+L
Sbjct: 123 QASDDWYRRAQLALQKGDEDLAREALKRRKSYADNASSLKAQLDQQKGVVENLVSNTRVL 182
Query: 188 ESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQAEAL 247
ESKI EA+ KKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVM MESQAEAL
Sbjct: 183 ESKIAEAKQKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMAMESQAEAL 242
Query: 248 GQLTSDDLEGKFAMLESSSVDDDLANLKKELSGSSKKGELPPGR---SSTRTGTPFRDAD 304
GQL +DDLEGKFA+LE+SSVDDDLA +KKE+SGSS KGELPPGR S++ PFRD +
Sbjct: 243 GQLATDDLEGKFALLETSSVDDDLAQMKKEISGSSSKGELPPGRTAVSNSGAARPFRDIE 302
Query: 305 IETELEQLRQRSKEF 319
IE EL +LR+++ E+
Sbjct: 303 IENELNELRKKANEY 317
>IM30_ARATH (O80796) Probable membrane-associated 30 kDa protein,
chloroplast precursor
Length = 330
Score = 411 bits (1057), Expect = e-114
Identities = 221/312 (70%), Positives = 266/312 (84%), Gaps = 7/312 (2%)
Query: 13 SAPLQSSSSSSSLR-KPLATSFFGTRPANTVKFRGMRIA--KPVRGGGAIGAGMNLFERF 69
S+P SS+ SLR PL TSFFG + ++ +R+A +R G GA MNLFERF
Sbjct: 21 SSPSTSSNRPCSLRILPLRTSFFGNS-SGALRVNVLRLACDNRLRCNGH-GATMNLFERF 78
Query: 70 TRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQA 129
+RVVKSYANA++SSFEDPEKILEQ V+EMN DLTKMRQATAQVLASQK+++NKYKAA Q+
Sbjct: 79 SRVVKSYANALISSFEDPEKILEQTVIEMNSDLTKMRQATAQVLASQKQLQNKYKAAQQS 138
Query: 130 SEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVSNTRLLES 189
S++WY++AQLAL KG+EDLAREALKRRKSFADNA+ALK QLDQQKGVVD+LVSNTRLLES
Sbjct: 139 SDDWYKRAQLALAKGDEDLAREALKRRKSFADNATALKTQLDQQKGVVDNLVSNTRLLES 198
Query: 190 KIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQAEALGQ 249
KIQEA++KKDTL ARA++AKT+TKV EM+G VNTSGALSAFEKMEEKVM MES+A+AL Q
Sbjct: 199 KIQEAKAKKDTLLARARTAKTATKVQEMIGTVNTSGALSAFEKMEEKVMAMESEADALTQ 258
Query: 250 LTSDDLEGKFAMLESSSVDDDLANLKKELSGSSKKGELPPGRSSTRTGT--PFRDADIET 307
+ +D+LEGKF MLE+SSVDDDLA+LKKELSGSSKKGELPPGRS+ T PF+D++IE
Sbjct: 259 IGTDELEGKFQMLETSSVDDDLADLKKELSGSSKKGELPPGRSTVAASTRYPFKDSEIEN 318
Query: 308 ELEQLRQRSKEF 319
EL +LR+++ +F
Sbjct: 319 ELNELRRKANDF 330
>Y617_SYNY3 (Q55707) Hypothetical protein sll0617
Length = 267
Score = 211 bits (536), Expect = 3e-54
Identities = 118/263 (44%), Positives = 175/263 (65%), Gaps = 10/263 (3%)
Query: 63 MNLFERFTRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENK 122
M LF+R RVV++ N +VS EDPEK+LEQAV++M +DL ++RQA A+ +A +KR E +
Sbjct: 1 MGLFDRLGRVVRANLNDLVSKAEDPEKVLEQAVIDMQEDLVQLRQAVARTIAEEKRTEQR 60
Query: 123 YKAATQASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVS 182
TQ +++W +A+LAL GEE+LAREAL R+KS D A+A + QL QQ+ + ++L
Sbjct: 61 LNQDTQEAKKWEDRAKLALTNGEENLAREALARKKSLTDTAAAYQTQLAQQRTMSENLRR 120
Query: 183 NTRLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMES 242
N LE+KI EA++KK+ L+ARA++AK + ++ + LG + TS A SAFE+ME KV+ ME+
Sbjct: 121 NLAALEAKISEAKTKKNMLQARAKAAKANAELQQTLGGLGTSSATSAFERMENKVLDMEA 180
Query: 243 QAEALGQLTSDDLEGKFAMLESSS-VDDDLANLKKELSGSSKKG--------ELPPGRSS 293
++A G+L +E +FA LE+SS V+D+LA LK ++G + G E P SS
Sbjct: 181 TSQAAGELAGFGIENQFAQLEASSGVEDELAALKASMAGGALPGTSAATPQLEAAPVDSS 240
Query: 294 TRTGTPFR-DADIETELEQLRQR 315
+ DA I+ EL+ LR+R
Sbjct: 241 VPANNASQDDAVIDQELDDLRRR 263
>PSPA_DEIRA (Q9RUB7) Phage shock protein A homolog
Length = 223
Score = 135 bits (341), Expect = 1e-31
Identities = 78/223 (34%), Positives = 128/223 (56%), Gaps = 2/223 (0%)
Query: 63 MNLFERFTRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENK 122
M++F+R +R++++ N ++S EDP KI++QA+ +M R A +A ++E +
Sbjct: 1 MSIFDRLSRLLRANVNDMISKAEDPAKIIDQALRDMRSAYADARNEVAGAMAQAAKLERE 60
Query: 123 YKAATQASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVS 182
++ + E+ +KA+ AL+ G EDLAREAL+R ++ D A Q Q+ VD L +
Sbjct: 61 AGTNSKLAAEYEKKAEEALRGGSEDLAREALRRAQNHKDLAKGFDEQRTVQQSTVDQLKT 120
Query: 183 NTRLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMES 242
R LE+KI E SKK L AR ++A+ + + G GA+ AF +ME+KV ME
Sbjct: 121 QLRALEAKIDEMESKKTLLAARQKTAQAGETLDRVSGFSKAGGAMDAFNEMEQKVAGMED 180
Query: 243 QAEALGQLTSD-DLEGKFAML-ESSSVDDDLANLKKELSGSSK 283
+ +A+G+L +D D + + L VDD LA LK ++ S++
Sbjct: 181 RNKAMGELRNDQDFDAQLKDLGRDKDVDDALAALKAKVQSSNQ 223
>PSPA_ECOLI (P23853) Phage shock protein A
Length = 221
Score = 116 bits (290), Expect = 8e-26
Identities = 65/215 (30%), Positives = 117/215 (54%), Gaps = 1/215 (0%)
Query: 65 LFERFTRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYK 124
+F RF +V + NA++ EDP+K++ + EM D L ++R +A+ LA +K++ + +
Sbjct: 2 IFSRFADIVNANINALLEKAEDPQKLVRLMIQEMEDTLVEVRSTSARALAEKKQLTRRIE 61
Query: 125 AATQASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVSNT 184
A+ EW KA+LAL K EDLAR AL ++ D +L+ ++ + +
Sbjct: 62 QASAREVEWQEKAELALLKEREDLARAALIEKQKLTDLIKSLEHEVTLVDDTLARMKKEI 121
Query: 185 RLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQA 244
LE+K+ E R+++ L R Q+A +S V L + A++ FE E ++ ME++A
Sbjct: 122 GELENKLSETRARQQALMLRHQAANSSRDVRRQLDSGKLDEAMARFESFERRIDQMEAEA 181
Query: 245 EALGQLTSDDLEGKFAMLES-SSVDDDLANLKKEL 278
E+ L+ +FA L++ ++ + LA LK ++
Sbjct: 182 ESHSFGKQKSLDDQFAELKADDAISEQLAQLKAKM 216
>PSPA_BACSU (P54617) Phage shock protein A homolog
Length = 226
Score = 108 bits (269), Expect = 2e-23
Identities = 75/229 (32%), Positives = 124/229 (53%), Gaps = 21/229 (9%)
Query: 64 NLFERFTRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKY 123
++ RF ++ + NA++ E+PEK+++Q + MN DL K++ TA V+A ++R + +Y
Sbjct: 1 SIIGRFKDIMSANINALLDKAENPEKMVDQYLRNMNSDLAKVKAETAAVMAEEQRAKREY 60
Query: 124 KAATQASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKA-------QLDQQKGV 176
E+ A ALQ G E AR+ L+R+ S S L+A Q + +
Sbjct: 61 HECQADMEKMESYAMKALQAGNESDARKFLERKTSLESKLSELQAANQIAATNAAQMRKM 120
Query: 177 VDSLVSNTRLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNV-NTSGALSAFEKMEE 235
D LVS+ I E ++K+ +KA+ AKT +++++ +V +TS ++SAF +ME+
Sbjct: 121 HDKLVSD-------IGELEARKNMIKAKWAVAKTQERMNKLGASVSSTSQSMSAFGRMED 173
Query: 236 KVMTMESQAEALGQLTS------DDLEGKFAMLESSSVDDDLANLKKEL 278
KV QA A+ +L S DL K+ SS VDD+LA LK ++
Sbjct: 174 KVNKALDQANAMAELNSAPQDDMADLSAKYDTGGSSQVDDELAALKAKM 222
>YVQH_BACSU (O32201) Hypothetical protein yvqH
Length = 225
Score = 87.0 bits (214), Expect = 6e-17
Identities = 55/197 (27%), Positives = 97/197 (48%), Gaps = 10/197 (5%)
Query: 81 VSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQASEEWYRKAQLA 140
+ E+P+ +L Q V +M D+ K +Q + + + KY+ A + + + +AQLA
Sbjct: 18 LDKMENPKVMLNQYVRDMESDIAKAKQTIVKQHTIAYQFKKKYEEAAEVAGKRKNQAQLA 77
Query: 141 LQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVSNTRLLESKIQEARSKKDT 200
GEE+LA++AL K A+ KA +Q + L LE+K+Q+ + KK
Sbjct: 78 FDAGEEELAKKALTEMKYLEGKAAEHKASYEQANSQLADLKEQLAALETKLQDVKDKKQA 137
Query: 201 LKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTME------SQAEALGQLT--- 251
L ARA +AK ++ +++ A F ++E ++ ME AEA +LT
Sbjct: 138 LIARANAAKAKEHMNTTFDKIDSESAYREFLRIENRIEEMEIRANYSKSAEAGTELTRKE 197
Query: 252 -SDDLEGKFAMLESSSV 267
+DD+E + + + S+
Sbjct: 198 FADDVEAEIEKMRTLSL 214
>MYSN_ACACA (P05659) Myosin II heavy chain, non muscle
Length = 1509
Score = 50.1 bits (118), Expect = 7e-06
Identities = 60/273 (21%), Positives = 119/273 (42%), Gaps = 46/273 (16%)
Query: 73 VKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQASEE 132
V+S N + +ED E ++ + +DL++ + T LA + + ++ + +E
Sbjct: 980 VESERNELQDKYED-EAAAHDSLKKKEEDLSRELRETKDALADAENISETLRSKLKNTER 1038
Query: 133 WYRKAQLALQKGEEDLAREAL---KRRKSFADNASALKAQLDQQKGVVDSLVSNTRLLES 189
+ L +D+ L K +KS + + +AQL+++K ++ S + L
Sbjct: 1039 GADDVRNEL----DDVTATKLQLEKTKKSLEEELAQTRAQLEEEKSGKEAASSKAKQLGQ 1094
Query: 190 KIQEARSKKDTLKARAQSAKTSTK--------VSEMLGNVNTSGALSAFEK--MEEKVMT 239
++++ARS+ D+LK++ +A+ S K + E L + T A +K +E K+
Sbjct: 1095 QLEDARSEVDSLKSKLSAAEKSLKTAKDQNRDLDEQLEDERTVRANVDKQKKALEAKLTE 1154
Query: 240 MESQAEAL-GQLTS-------------------DDLEGKFAMLESSSVD--DDLANLKKE 277
+E Q AL GQ + ++ E A LE + D++A L +
Sbjct: 1155 LEDQVTALDGQKNAAAAQAKTLKTQVDETKRRLEEAEASAARLEKERKNALDEVAQLTAD 1214
Query: 278 LSGSSKKGELPPGRSSTRTGTPFRDADIETELE 310
L G + +TR +++++ELE
Sbjct: 1215 LDAERDSGAQQRRKLNTRI------SELQSELE 1241
>RA50_METKA (Q8TXI4) DNA double-strand break repair rad50 ATPase
Length = 876
Score = 49.7 bits (117), Expect = 1e-05
Identities = 63/254 (24%), Positives = 114/254 (44%), Gaps = 42/254 (16%)
Query: 90 ILEQAVLEMNDDLTKMRQATAQVLASQKRMENK------YKAATQASEEWYRKAQLALQ- 142
+ +AV ++ K+ +AT + +KR+ ++ +K A + + E R A+ L+
Sbjct: 130 VFREAVYIRQGEIAKLVEATRE---ERKRIVDRTLGLAEFKKAREQAHELLRVAEAKLET 186
Query: 143 --------KG-EEDLAR-----EALKRR-KSFADNASALKAQLDQ---QKGVVDSLVSNT 184
KG +++L R E LKR K LK +L++ K + L
Sbjct: 187 FRERVRDLKGSKKELKRVERELEELKREVKELEPEVEELKERLNELREAKREFERLEGEL 246
Query: 185 RLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQA 244
RLLE+KI+ + ++D L+ + K + + + LG+V S ++E + + +
Sbjct: 247 RLLENKIESLKGRRDDLRKLVEEGKEAERELQRLGDVP-----SKVRELENEEAELRRRI 301
Query: 245 EALGQLTSDDLEGKFAMLESSSVDDDLANLKKELSGSSKKGELPPGRSSTRTGTPFRDAD 304
E L L DDL LES+ +++L +K+EL + + P R F+D
Sbjct: 302 EELRNLL-DDLRSLRNRLESA--EEELEGVKRELEELKDEAGVDPERL-----VEFKDKI 353
Query: 305 IETELEQLRQRSKE 318
+E E+LR +E
Sbjct: 354 VEAS-ERLRDLRRE 366
Score = 32.0 bits (71), Expect = 2.1
Identities = 44/195 (22%), Positives = 77/195 (38%), Gaps = 29/195 (14%)
Query: 82 SSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQASEEWYRKAQ--- 138
S ED E+ L + E + K+R+A + L +E K K A +A +E R +
Sbjct: 575 SGVEDVEEELRRLEEERDHVGQKLREAEGE-LERYHNLEEKVKRAREARKELKRIERDLE 633
Query: 139 ---------------LALQKGEEDLAREAL----KRRKSFADNASALKAQLDQQKGVVDS 179
L + G ED E L K+ + D S +K +L+ + +
Sbjct: 634 DAKGRLEQVERNLEGLRERYGSEDRLEEELESVEKKYERVRDKLSEVKGRLNGMEKRREE 693
Query: 180 LVSNTRLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMT 239
L R K +EA+ +K+ L+ + +V +V L A E+ K+
Sbjct: 694 LKKQVR----KYREAKERKERLERVVEVLSLCKEVFRYSRDVAREKVLPAVEREASKI-- 747
Query: 240 MESQAEALGQLTSDD 254
++ ++ G L +D
Sbjct: 748 LQDLSDRYGSLRIED 762
>M21_STRPY (P50468) M protein, serotype 2.1 precursor
Length = 407
Score = 47.8 bits (112), Expect = 4e-05
Identities = 62/270 (22%), Positives = 119/270 (43%), Gaps = 27/270 (10%)
Query: 65 LFERFTRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTK--MRQATAQVLASQKRMENK 122
LFE+ +V + + +D EK+ ++ ++D+ + +RQ + Q+R +N
Sbjct: 74 LFEKLDKVEEEHKKVEEEHKKDHEKLEKK-----SEDVERHYLRQLDQEYKEQQERQKNL 128
Query: 123 YKAATQASEEWYRKAQLALQKGEE--------DLAREALKRRKSFADNASALKAQLDQQK 174
+ Q+ E ++ Q LQK ++ + +R++L RR A A+ + + QK
Sbjct: 129 EELERQSQREVEKRYQEQLQKQQQLEKEKQISEASRKSL-RRDLEASRAAKKDLEAEHQK 187
Query: 175 GVVDSLVS--NTRLLESKIQEARSKKDTLKARAQSAKTSTKVSE-----MLGNVNTSGAL 227
+ +S + + L ++ +R+ K L+A Q K ++SE + ++ S A
Sbjct: 188 LKEEKQISEASRKSLRRDLEASRAAKKDLEAEHQKLKEEKQISEASRQGLSRDLEASRAA 247
Query: 228 SAFEKMEEKVMTMESQ-AEALGQLTSDDLEGKFAMLESSSVDDDLANLKKELSGSSK-KG 285
+ E + + E Q +EA Q S DLE + V+ DLA +L K
Sbjct: 248 KKDLEAEHQKLKEEKQISEASRQGLSRDLEA--SREAKKKVEADLAEANSKLQALEKLNK 305
Query: 286 ELPPGRSSTRTGTPFRDADIETELEQLRQR 315
EL G+ + A +E E + L+++
Sbjct: 306 ELEEGKKLSEKEKAELQAKLEAEAKALKEQ 335
>35KD_MYCTU (P31511) 35 kDa protein
Length = 270
Score = 47.4 bits (111), Expect = 5e-05
Identities = 49/257 (19%), Positives = 106/257 (41%), Gaps = 26/257 (10%)
Query: 64 NLFERFTRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKY 123
N F + + + + ++ + DP+ ++QA+ E + Q AQV+ +Q+++E +
Sbjct: 3 NPFVKAWKYLMALFSSKIDEHADPKVQIQQAIEEAQRTHQALTQQAAQVIGNQRQLEMRL 62
Query: 124 KAATQASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVS- 182
E+ + AL ++ A + + + A A AQL + V+ L +
Sbjct: 63 NRQLADIEKLQVNVRQALTLADQATAAGDAAKATEYNNAAEAFAAQLVTAEQSVEDLKTL 122
Query: 183 -----------------NTRLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVN--- 222
N +L+ KI E + L ++ + AK +VS L +++
Sbjct: 123 HDQALSAAAQAKKAVERNAMVLQQKIAE----RTKLLSQLEQAKMQEQVSASLRSMSELA 178
Query: 223 TSGALSAFEKMEEKVMTMESQAEALGQLTSDDLEGKFAMLESSSVDDDLANLKKELSGSS 282
G + +++ +K+ + A +L ++G+ +E + + + + E +S
Sbjct: 179 APGNTPSLDEVRDKIERRYANAIGSAELAESSVQGRMLEVEQAGI-QMAGHSRLEQIRAS 237
Query: 283 KKGELPPGRSSTRTGTP 299
+GE P +T T P
Sbjct: 238 MRGEALPAGGTTATPRP 254
>RRB1_MOUSE (Q99PL5) Ribosome-binding protein 1 (Ribosome receptor
protein) (mRRp)
Length = 1605
Score = 47.0 bits (110), Expect = 6e-05
Identities = 49/194 (25%), Positives = 98/194 (50%), Gaps = 16/194 (8%)
Query: 91 LEQAVLEMNDDLTKMRQATAQVLASQKRMENK---YKAATQASEEWYRKAQLALQKGEED 147
L Q + ++N +L + +A+ Q +K +E K ++ + +++++ ALQK E+
Sbjct: 1030 LRQELSKVNKELVEKSEASRQEEQQRKALEAKAATFEKQVLQLQASHKESEEALQKRLEE 1089
Query: 148 LAREALKRRKSFAD-NASALKAQLDQQK--GVVDSLVSNTRLLESKIQEARSKKDTLK-A 203
+ RE + + S A+ A A KAQ QQ+ + L S+ ++SK +E S LK A
Sbjct: 1090 VTRELCRAQTSHANLRADAEKAQEQQQRVAELHSKLQSSEVEVKSKCEELSSLHGQLKEA 1149
Query: 204 RAQSAKTSTKVSEMLGNV------NTSGALSAFEKMEEKVMTMESQAEALGQLTSDDLEG 257
RA++++ + ++ + + +T + + + + ++ +ESQ L + TS E
Sbjct: 1150 RAENSQLTERIRSIEALLEAGQAQDTQASHAEANQQQTRLKELESQVSCLEKETS---EL 1206
Query: 258 KFAMLESSSVDDDL 271
K AM + ++DL
Sbjct: 1207 KEAMEQQKGKNNDL 1220
Score = 34.3 bits (77), Expect = 0.42
Identities = 38/184 (20%), Positives = 83/184 (44%), Gaps = 27/184 (14%)
Query: 161 DNASALKAQLDQQKGVVDSLVSNTRLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGN 220
D + LK QL +++ ++ + + + +SK++E + + KA+A + + K +++
Sbjct: 896 DPVAILKRQLQEKEKLLATEQEDAAVAKSKLRELNKEMASEKAKAAAGEAKVK-KQLVAR 954
Query: 221 VNTSGALSA------------FEKMEEKVMTMESQAE-----ALGQLTSDDLEGKFAMLE 263
A+ A ++++ K+ T++ Q E L +L ++ + A+ +
Sbjct: 955 EQEIAAVQARMQASYRDHVKEVQQLQGKIRTLQEQLENGPNTQLARLQQENSILRDALNQ 1014
Query: 264 SSS-----VDDDLANLKKELSGSSK----KGELPPGRSSTRTGTPFRDADIETELEQLRQ 314
++S + +LA L++ELS +K K E R + A E ++ QL+
Sbjct: 1015 ATSQVESKQNTELAKLRQELSKVNKELVEKSEASRQEEQQRKALEAKAATFEKQVLQLQA 1074
Query: 315 RSKE 318
KE
Sbjct: 1075 SHKE 1078
Score = 30.0 bits (66), Expect = 8.0
Identities = 39/201 (19%), Positives = 82/201 (40%), Gaps = 20/201 (9%)
Query: 73 VKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQASEE 132
++S + + + ++ +EQ + ND K +A + +++ E K ++ TQA EE
Sbjct: 1192 LESQVSCLEKETSELKEAMEQQKGKNNDLREKNWKAMEALALAERACEEKLRSLTQAKEE 1251
Query: 133 WYRKAQLALQKGEEDLAR----EALKRRKSFADNASALKAQLDQQKGVVDSLVSNTRLLE 188
++ LA + +E L ++ +++A+ K + + +L + ++
Sbjct: 1252 SEKQLHLAEAQTKETLLALLPGLSISAHQNYAEWLQEFKEKGSELLKKPPTLEPSMDIV- 1310
Query: 189 SKIQEARSKKDTLKARAQSAKT--------------STKVSEMLGNVNTSGALSAFEKME 234
K++EA +++L+A +T S + E + A K
Sbjct: 1311 LKLREAEETQNSLQAECDQYRTILAETEGMLKDLQKSVEEEERVWKAKVGAAEEELHKSR 1370
Query: 235 EKVMTMESQAEAL-GQLTSDD 254
V +E E L G+L S D
Sbjct: 1371 VTVKHLEDIVEKLKGELESSD 1391
>TRIP_HUMAN (Q9BWF2) TRAF-interacting protein
Length = 469
Score = 45.1 bits (105), Expect = 2e-04
Identities = 48/213 (22%), Positives = 95/213 (44%), Gaps = 17/213 (7%)
Query: 83 SFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQASEEWYRKAQLALQ 142
S +D EK Q +++ D + R AT V++ Q+ + KA S + L Q
Sbjct: 91 SQKDKEKRDSQVIIDTLRDTLEERNAT--VVSLQQALG---KAEMLCSTLKKQMKYLEQQ 145
Query: 143 KGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVSNTRLLESKIQEARSKKDTLK 202
+ E A+E +R +S ++ L Q+ V+ ++ + + +S +++ +LK
Sbjct: 146 QDETKQAQEEARRLRSKMKTMEQIELLLQSQRPEVEEMIRDMGVGQSAVEQLAVYCVSLK 205
Query: 203 ARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQAEAL-GQLTSDDLEGKFAM 261
++ K + K S + +K+ + + + S+ + + +L LE K A
Sbjct: 206 KEYENLKEARKASGEVA-----------DKLRKDLFSSRSKLQTVYSELDQAKLELKSAQ 254
Query: 262 LESSSVDDDLANLKKELSGSSKKGELPPGRSST 294
+ S D ++ +LKK+L+ + LPP S T
Sbjct: 255 KDLQSADKEIMSLKKKLTMLQETLNLPPVASET 287
>SLA2_YEAST (P33338) SLA2 protein (Transmembrane protein MOP2)
Length = 968
Score = 44.3 bits (103), Expect = 4e-04
Identities = 51/244 (20%), Positives = 99/244 (39%), Gaps = 22/244 (9%)
Query: 90 ILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQASEEWYRKAQLALQKGEEDLA 149
I QA +M D +QA Q Q R+E + Q + Q LQK ++D+
Sbjct: 347 IFPQATAQMQPDFWANQQA--QFANEQNRLEQERVQQLQQQQAQQELFQQQLQKAQQDMM 404
Query: 150 REALKRRKSFADNASALKAQLDQQKGVVDSLVSNTRLLESKI--------QEARSKKDTL 201
L+++ ++ AL Q ++ + ++ + LES+I ++ +K + L
Sbjct: 405 NMQLQQQNQHQNDLIALTNQYEKDQALLQQYDQRVQQLESEITTMDSTASKQLANKDEQL 464
Query: 202 KARAQSAKTSTKVSEMLGNVNTS------GALSAFEKMEEKVMTMESQAEALGQLTSDDL 255
A + E L + + L F+K++ KV + + + QL L
Sbjct: 465 TALQDQLDVWERKYESLAKLYSQLRQEHLNLLPRFKKLQLKVNSAQESIQKKEQL-EHKL 523
Query: 256 EGKFAMLESSSVDDDLANLKKELSGSSKKGELPPGRSSTRTGT-----PFRDADIETELE 310
+ K + D D A L+ E S ++ + + ++ T T P DA +E+ +
Sbjct: 524 KQKDLQMAELVKDRDRARLELERSINNAEADSAAATAAAETMTQDKMNPILDAILESGIN 583
Query: 311 QLRQ 314
+++
Sbjct: 584 TIQE 587
>REST_HUMAN (P30622) Restin (Cytoplasmic linker protein-170 alpha-2)
(CLIP-170) (Reed-Sternberg intermediate filament
associated protein)
Length = 1427
Score = 43.1 bits (100), Expect = 0.001
Identities = 52/233 (22%), Positives = 101/233 (43%), Gaps = 26/233 (11%)
Query: 68 RFTRVVKSYANAIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQV---------LASQKR 118
+ +V+K N++ + +K +Q ++EM D L K+++A +V Q +
Sbjct: 687 KLMKVIKEKENSLEAIRSKLDKAEDQHLVEMEDTLNKLQEAEIKVKELEVLQAKCNEQTK 746
Query: 119 MENKYKAATQASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVD 178
+ + + + +A+EE AL+K + E K R+ +K L+ +K
Sbjct: 747 VIDNFTSQLKATEEKLLDLD-ALRKASSEGKSEMKKLRQQLEAAEKQIK-HLEIEKNAES 804
Query: 179 SLVSN-TRLLESKIQEARSKKDTLKARAQSAKTSTKVSEMLGN---VNTSGALSAFEKME 234
S S+ TR L+ + + + ++ L +Q +T K ++L + A+S M+
Sbjct: 805 SKASSITRELQGRELKLTNLQENLSEVSQVKETLEKELQILKEKFAEASEEAVSVQRSMQ 864
Query: 235 EKVMTMESQAEALGQLTSD---------DLEGKFAMLESSSVDDDLANLKKEL 278
E V + + E L+SD D+E KF E ++ L K++L
Sbjct: 865 ETVNKLHQKEEQFNMLSSDLEKLRENLADMEAKFR--EKDEREEQLIKAKEKL 915
Score = 34.7 bits (78), Expect = 0.32
Identities = 47/218 (21%), Positives = 90/218 (40%), Gaps = 25/218 (11%)
Query: 95 VLEMNDDLTKMRQATAQVLASQKRMENKYKAATQASEEWYRKAQLALQKGEEDLAREALK 154
V E+N + ++ +K +E +AA Q S++ ALQ+ LA E +
Sbjct: 1123 VEELNKSKELLTVENQKMEEFRKEIETLKQAAAQKSQQLS-----ALQEENVKLAEELGR 1177
Query: 155 RRKSFADNASALKAQLDQQKGVVDSLVSNTRLLESKIQEARSKKDTLKARAQSAKTSTKV 214
R + +L++++ V+ N +LLE K +E++ KD + +A K+ +
Sbjct: 1178 SRDEVTSHQ-----KLEEERSVL-----NNQLLEMKKRESKFIKDADEEKASLQKSISIT 1227
Query: 215 SEMLGNVNTSGALSAFEKMEEKVMTMESQAEALGQLTSDDLEGKFAMLESSSVDDDLANL 274
S +L + + EK+ +V + + ++ L LES V +L
Sbjct: 1228 SALLTEKD-----AELEKLRNEVTVLRGE-----NASAKSLHSVVQTLESDKVKLELKVK 1277
Query: 275 KKELSGSSKKGELPPGRSSTRTGTPFRDADIETELEQL 312
EL K +L +T T + E++++ L
Sbjct: 1278 NLELQLKENKRQLSSSSGNTDTQADEDERAQESQIDFL 1315
>MYH7_HUMAN (P12883) Myosin heavy chain, cardiac muscle beta isoform
(MyHC-beta)
Length = 1935
Score = 43.1 bits (100), Expect = 0.001
Identities = 68/303 (22%), Positives = 116/303 (37%), Gaps = 65/303 (21%)
Query: 79 AIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYK----------AATQ 128
A+ S + K L+ + E+ ++L R A A+V + + + + AT
Sbjct: 1099 ALGSQLQKKLKELQARIEELEEELESERTARAKVEKLRSDLSRELEEISERLEEAGGATS 1158
Query: 129 ASEEWYRKAQLALQKGEEDLAREALKR-------RKSFADNASALKAQLDQQKGV----- 176
E +K + QK DL L+ RK AD+ + L Q+D + V
Sbjct: 1159 VQIEMNKKREAEFQKMRRDLEEATLQHEATAAALRKKHADSVAELGEQIDNLQRVKQKLE 1218
Query: 177 ---------VDSLVSN--------------TRLLESKIQEARSKKDTLK-----ARAQSA 208
+D + SN R LE ++ E RSK + + +Q A
Sbjct: 1219 KEKSEFKLELDDVTSNMEQIIKAKANLEKMCRTLEDQMNEHRSKAEETQRSVNDLTSQRA 1278
Query: 209 KTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQAEALGQLTSDDLEGKFAM---LESS 265
K T+ E+ ++ AL ++ +T Q E L + ++++ K A+ L+S+
Sbjct: 1279 KLQTENGELSRQLDEKEAL--ISQLTRGKLTYTQQLEDLKRQLEEEVKAKNALAHALQSA 1336
Query: 266 SVDDDL--------ANLKKELSG--SSKKGELPPGRSSTRTGTPFRDADIETELEQLRQR 315
D DL K EL S E+ R+ T R ++E ++L QR
Sbjct: 1337 RHDCDLLREQYEEETEAKAELQRVLSKANSEVAQWRTKYETDAIQRTEELEEAKKKLAQR 1396
Query: 316 SKE 318
+E
Sbjct: 1397 LQE 1399
Score = 33.5 bits (75), Expect = 0.72
Identities = 51/252 (20%), Positives = 104/252 (41%), Gaps = 38/252 (15%)
Query: 91 LEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQ-----------------ASEEW 133
LE E+ D+ + A+V + ENK K T+ A +E
Sbjct: 943 LEDECSELKRDIDDLELTLAKVEKEKHATENKVKNLTEEMAGLDEIIAKLTKEKKALQEA 1002
Query: 134 YRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGV-------VDSLVSNTRL 186
+++A LQ EED K + L+ L+Q+K V L + +L
Sbjct: 1003 HQQALDDLQ-AEEDKVNTLTKAKVKLEQQVDDLEGSLEQEKKVRMDLERAKRKLEGDLKL 1061
Query: 187 LESKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQAEA 246
+ I + + K L R + K +++ + + AL + ++++K+ ++++ E
Sbjct: 1062 TQESIMDLENDKQQLDERLK--KKDFELNALNARIEDEQALGS--QLQKKLKELQARIEE 1117
Query: 247 LGQLTSDDLEGKFAMLESSSVDDDLANLKKELSGSSKKGELPPGRSSTRTGTPFRDADIE 306
L ++LE + + V+ ++L +EL S++ E G +S + + E
Sbjct: 1118 L----EEELESE--RTARAKVEKLRSDLSRELEEISERLEEAGGATSVQIE---MNKKRE 1168
Query: 307 TELEQLRQRSKE 318
E +++R+ +E
Sbjct: 1169 AEFQKMRRDLEE 1180
Score = 32.7 bits (73), Expect = 1.2
Identities = 38/206 (18%), Positives = 84/206 (40%), Gaps = 31/206 (15%)
Query: 79 AIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRM-------ENKYKAATQASE 131
A+V E K+ EQ ++E ++ + + ++ +K+M + + + A Q
Sbjct: 1690 AVVEQTERSRKLAEQELIETSERVQLLHSQNTSLINQKKKMDADLSQLQTEVEEAVQECR 1749
Query: 132 EWYRKAQLALQ---------KGEEDLAREALKRRKSFADNASALKAQLDQQKGV------ 176
KA+ A+ K E+D + + +K+ L+ +LD+ + +
Sbjct: 1750 NAEEKAKKAITDAAMMAEELKKEQDTSAHLERMKKNMEQTIKDLQHRLDEAEQIALKGGK 1809
Query: 177 --VDSLVSNTRLLESKIQEARSKKDTLKARAQSAKTSTKVSEML-----GNVNTSGALSA 229
+ L + R LE+++ EA K++ + K+ ++ E+ N
Sbjct: 1810 KQLQKLEARVRELENEL-EAEQKRNAESVKGM-RKSERRIKELTYQTEEDRKNLLRLQDL 1867
Query: 230 FEKMEEKVMTMESQAEALGQLTSDDL 255
+K++ KV + QAE + + +L
Sbjct: 1868 VDKLQLKVKAYKRQAEEAEEQANTNL 1893
>SMC2_CHICK (Q90988) Structural maintenance of chromosome 2
(Chromosome scaffold protein ScII)
Length = 1189
Score = 42.7 bits (99), Expect = 0.001
Identities = 53/224 (23%), Positives = 93/224 (40%), Gaps = 35/224 (15%)
Query: 103 TKMRQATAQVLASQKRMENKYKAATQASEEWYRKAQL--------ALQKGEEDLAREALK 154
T +AT + LA+ K M KY+ Q E +A+L A K +EDL L
Sbjct: 692 TSQLEATEKELANLKNMAEKYQHLKQQWEMKSEEAELLQTKIQQSAYHKQQEDL----LA 747
Query: 155 RRKSFADNASALKAQLDQQKGVVDSLVSNTRLLESKIQEARSKKDTLKARAQSAKTSTKV 214
+K+ A+ LK + Q+ + + LE+K++ A +++ AQ S K
Sbjct: 748 LKKTIAECEETLKKTEESQRKAEEEY----KALENKMKNAEAERGKEIKNAQQKLNSAK- 802
Query: 215 SEMLGNVNTSGALSAFEKMEEKVMTMESQAEALGQLTSDDLEGKFAMLESSSVDDDLANL 274
A + KM+EK +E+ L QL + K +S + +A+L
Sbjct: 803 ---------KKADDSSRKMKEKQQEVEALVLELEQLKQEQASYK---QQSEAAQQAIASL 850
Query: 275 KKELSGSSKKGELPPGRSSTRTGTPFRDADIETELEQLRQRSKE 318
K+++S L TR + ++ +E + +R+K+
Sbjct: 851 KEQVSA------LEAEAVKTRESLKNAENELSSEKGLMEERTKD 888
Score = 33.9 bits (76), Expect = 0.55
Identities = 47/239 (19%), Positives = 90/239 (36%), Gaps = 20/239 (8%)
Query: 82 SSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKA-------------ATQ 128
+S++ + +QA+ + + ++ + + S K EN+ + A
Sbjct: 834 ASYKQQSEAAQQAIASLKEQVSALEAEAVKTRESLKNAENELSSEKGLMEERTKDIKAKS 893
Query: 129 ASEEWYRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVSNTRLLE 188
A E YR+ LQ L + K ++ AD +S L L + K + +
Sbjct: 894 AKIEKYREQNNELQLSINALEHDINKYQQETADASSTLDKLLKEYKWIASEKELFGQADT 953
Query: 189 SKIQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEK-----MEEKVMTMESQ 243
+ EA + K+T + Q T + E N+ LS E+ M++K M +
Sbjct: 954 TYDFEANNPKET-GQKLQKLLTKKEKLEKSLNMRAMNLLSEAEERYNDLMKKKRMVENDK 1012
Query: 244 AEALGQLTS-DDLEGKFAMLESSSVDDDLANLKKELSGSSKKGELPPGRSSTRTGTPFR 301
+ L + D + K + V+ D ++ L +K +P + + G FR
Sbjct: 1013 IKILATIEELDRKKNKALHIAWEKVNKDFGSIFSMLLPGAKAMLVPSKKQNILDGLEFR 1071
>MYSS_CYPCA (Q90339) Myosin heavy chain, fast skeletal muscle
Length = 1935
Score = 42.7 bits (99), Expect = 0.001
Identities = 54/249 (21%), Positives = 108/249 (42%), Gaps = 32/249 (12%)
Query: 91 LEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQ--ASEEW----YRKAQLALQKG 144
LE E+ D+ + A+V + ENK K T+ AS++ K + ALQ+
Sbjct: 944 LEDECSELKKDIDDLELTLAKVEKEKHATENKVKNLTEEMASQDESIAKLTKEKKALQEA 1003
Query: 145 ----------EEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVSNTRLLESKIQEA 194
EED K + L+ L+Q+K + L R LE ++ A
Sbjct: 1004 HQQTLDDLQAEEDKVNTLTKAKTKLEQQVDDLEGSLEQEKKLRMDLERAKRKLEGDLKLA 1063
Query: 195 RSKKDTLKARAQSA-----KTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQAEALGQ 249
+ L+ Q + K ++S++L + +L A ++++K+ ++++ E L
Sbjct: 1064 QESIMDLENEKQQSDEKIKKKDFEISQLLSKIEDEQSLGA--QLQKKIKELQARIEEL-- 1119
Query: 250 LTSDDLEGKFAMLESSSVDDDLANLKKELSGSSKKGELPPGRSSTRTGTPFRDADIETEL 309
+++E + A + V+ A+L +EL S++ E G ++ + + E E
Sbjct: 1120 --EEEIEAERA--ARAKVEKQRADLSRELEEISERLEEAGGATAAQIE---MNKKREAEF 1172
Query: 310 EQLRQRSKE 318
+++R+ +E
Sbjct: 1173 QKMRRDLEE 1181
>MYH4_RABIT (Q28641) Myosin heavy chain, skeletal muscle, juvenile
Length = 1938
Score = 42.7 bits (99), Expect = 0.001
Identities = 41/175 (23%), Positives = 79/175 (44%), Gaps = 16/175 (9%)
Query: 80 IVSSFEDPEKILEQAVLE---MNDDLTKMRQATAQVLASQKRMENKYKAATQASEEWYRK 136
IV + E+ ++A+ + M ++L K + +A + +K ME K +E
Sbjct: 1747 IVQEARNAEEKAKKAITDAAMMAEELKKEQDTSAHLERMKKNMEQTVKDLQHRLDE---A 1803
Query: 137 AQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGV------VDSLVSNTRLLESK 190
QLAL+ G++ + + + R+ A+ S K ++ KG+ V L T
Sbjct: 1804 EQLALKGGKKQIQKLEARVRELEAEVESEQKRNVEAVKGLRKHERRVKELTYQTEEDRKN 1863
Query: 191 IQEARSKKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQAE 245
+ + D L+A+ +S K + +E N+N LS F K++ ++ E +A+
Sbjct: 1864 VLRLQDLVDKLQAKVKSYKRQAEEAEEQCNIN----LSKFRKLQHELEEAEERAD 1914
Score = 38.5 bits (88), Expect = 0.022
Identities = 59/303 (19%), Positives = 114/303 (37%), Gaps = 65/303 (21%)
Query: 79 AIVSSFEDPEKILEQAVLEMNDDLTKMRQATAQVLASQKRMENKYK----------AATQ 128
A+ + K L+ + E+ +++ R + A+ + + + + AT
Sbjct: 1102 ALAMQLQKKIKELQARIEELEEEIEAERASRAKAEKQRSDLSRELEEISERLEEAGGATS 1161
Query: 129 ASEEWYRKAQLALQKGEEDLAREALKR-------RKSFADNASALKAQLDQQKGV----- 176
A E +K + QK DL L+ RK AD+ + L Q+D + V
Sbjct: 1162 AQIEMNKKREAEFQKMRRDLEEATLQHEATAATLRKKHADSVAELGEQIDNLQRVKQKLE 1221
Query: 177 ---------VDSLVSN--------------TRLLESKIQEARSKKDTLK-----ARAQSA 208
+D L SN R LE ++ E ++K++ + AQ A
Sbjct: 1222 KEKSELKMEIDDLASNMETVSKAKGNLEKMCRTLEDQVSELKTKEEEHQRLINDLSAQRA 1281
Query: 209 KTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQAEALGQLTSDDLEGKFAM---LESS 265
+ T+ E ++ +L ++ Q E L + ++++ K A+ L+S+
Sbjct: 1282 RLQTESGEFSRQLDEKDSL--VSQLSRGKQAFTQQIEELKRQLEEEIKAKSALAHALQSA 1339
Query: 266 SVDDDL----------ANLKKELSGSSKKGELPPGRSSTRTGTPFRDADIETELEQLRQR 315
D DL A + + + S E+ R+ T R ++E ++L QR
Sbjct: 1340 RHDCDLLREQYEEEQEAKAELQRAMSKANSEVAQWRTKYETDAIQRTEELEEAKKKLAQR 1399
Query: 316 SKE 318
++
Sbjct: 1400 LQD 1402
Score = 35.8 bits (81), Expect = 0.15
Identities = 49/250 (19%), Positives = 103/250 (40%), Gaps = 34/250 (13%)
Query: 91 LEQAVLEMNDDLTKMRQATAQVLASQKRMENKYKAATQ-----------------ASEEW 133
LE E+ D+ + A+V + ENK K T+ A +E
Sbjct: 946 LEDECSELKKDIDDLELTLAKVEKEKHATENKVKNLTEEMAGLDETIAKLTKEKKALQEA 1005
Query: 134 YRKAQLALQKGEEDLAREALKRRKSFADNASALKAQLDQQKGVVDSLVSNTRLLESKIQE 193
+++ LQ EED K + L+ L+Q+K + L R LE ++
Sbjct: 1006 HQQTLDDLQ-AEEDKVNTLTKAKTKLEQQVDDLEGSLEQEKKIRMDLERAKRKLEGDLKL 1064
Query: 194 ARS-----KKDTLKARAQSAKTSTKVSEMLGNVNTSGALSAFEKMEEKVMTMESQAEALG 248
A+ + D + + K ++S + + AL+ ++++K+ ++++ E L
Sbjct: 1065 AQESTMDIENDKQQLDEKLKKKEFEMSNLQSKIEDEQALAM--QLQKKIKELQARIEEL- 1121
Query: 249 QLTSDDLEGKFAMLESSSVDDDLANLKKELSGSSKKGELPPGRSSTRTGTPFRDADIETE 308
+++E + A + + ++L +EL S++ E G +S + + E E
Sbjct: 1122 ---EEEIEAERA--SRAKAEKQRSDLSRELEEISERLEEAGGATSAQIE---MNKKREAE 1173
Query: 309 LEQLRQRSKE 318
+++R+ +E
Sbjct: 1174 FQKMRRDLEE 1183
Database: sprot
Posted date: Nov 25, 2004 10:54 AM
Number of letters in database: 59,974,054
Number of sequences in database: 164,201
Lambda K H
0.309 0.123 0.316
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 29,951,788
Number of Sequences: 164201
Number of extensions: 1125916
Number of successful extensions: 5044
Number of sequences better than 10.0: 434
Number of HSP's better than 10.0 without gapping: 26
Number of HSP's successfully gapped in prelim test: 408
Number of HSP's that attempted gapping in prelim test: 4659
Number of HSP's gapped (non-prelim): 730
length of query: 319
length of database: 59,974,054
effective HSP length: 110
effective length of query: 209
effective length of database: 41,911,944
effective search space: 8759596296
effective search space used: 8759596296
T: 11
A: 40
X1: 16 ( 7.1 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 42 (21.7 bits)
S2: 66 (30.0 bits)
Medicago: description of AC137825.9