
BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
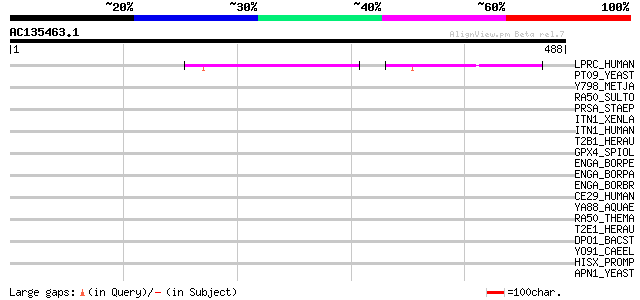
Query= AC135463.1 - phase: 0 /pseudo
(488 letters)
Database: sprot
164,201 sequences; 59,974,054 total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
LPRC_HUMAN (P42704) 130 kDa leucine-rich protein (LRP 130) (GP13... 52 3e-06
PT09_YEAST (P32522) PET309 protein, mitochondrial precursor 39 0.030
Y798_METJA (Q58208) Hypothetical protein MJ0798 38 0.052
RA50_SULTO (Q96YR5) DNA double-strand break repair rad50 ATPase 35 0.33
PRSA_STAEP (Q8CNR4) Foldase protein prsA precursor (EC 5.2.1.8) 35 0.44
ITN1_XENLA (O42287) Intersectin 1 35 0.44
ITN1_HUMAN (Q15811) Intersectin 1 (SH3 domain-containing protein... 35 0.57
T2B1_HERAU (P25257) Type II restriction enzyme HgiBI (EC 3.1.21.... 34 0.97
GPX4_SPIOL (O23814) Probable phospholipid hydroperoxide glutathi... 34 0.97
ENGA_BORPE (Q7VWL4) GTP-binding protein engA 34 0.97
ENGA_BORPA (Q7W6Q0) GTP-binding protein engA 34 0.97
ENGA_BORBR (Q7WHN4) GTP-binding protein engA 34 0.97
CE29_HUMAN (O15078) Centrosomal protein Cep290 34 0.97
YA88_AQUAE (O67178) Hypothetical protein AQ_1088 33 1.3
RA50_THEMA (Q9X1X1) Probable DNA double-strand break repair rad5... 33 1.3
T2E1_HERAU (P25260) Type II restriction enzyme HgiEI (EC 3.1.21.... 33 1.7
DPO1_BACST (P52026) DNA polymerase I (EC 2.7.7.7) (POL I) 33 1.7
YO91_CAEEL (P41842) Hypothetical protein T20B12.1 in chromosome III 33 2.2
HISX_PROMP (Q7V004) Histidinol dehydrogenase (EC 1.1.1.23) (HDH) 33 2.2
APN1_YEAST (P22936) DNA-(apurinic or apyrimidinic site) lyase 1 ... 33 2.2
>LPRC_HUMAN (P42704) 130 kDa leucine-rich protein (LRP 130) (GP130)
(Leucine-rich PPR-motif containing protein)
Length = 1273
Score = 52.4 bits (124), Expect = 3e-06
Identities = 46/162 (28%), Positives = 73/162 (44%), Gaps = 8/162 (4%)
Query: 154 LANCASLENLRKTEET-------FNKMRELGFPVTAFACNQLLLIYKKIDKK-KIADVLL 205
L +C SL K EE ++ +++LG N LL +Y + + K D L
Sbjct: 6 LRSCGSLLPELKLEERTEFAHRIWDTLQKLGAVYDVSHYNALLKVYLQNEYKFSPTDFLA 65
Query: 206 MMEKENVKPSSYTYKILIDVKGLSNDIDGMSQIVETMKAEGCELDHLTRASLARHYAAAG 265
ME+ N++P+ TY+ LI DI+G S+I+ MK + + ++L +A AG
Sbjct: 66 KMEEANIQPNRVTYQRLIASYCNVGDIEGASKILGFMKTKDLPVTEAVFSALVTGHARAG 125
Query: 266 LTEKTEAILKEIEGENLKENMWVCPTLLRLYAILGRADEVER 307
E E IL + ++ LL YA G D V++
Sbjct: 126 DMENAENILTVMRDAGIEPGPDTYLALLNAYAEKGDIDHVKQ 167
Score = 49.3 bits (116), Expect = 2e-05
Identities = 37/143 (25%), Positives = 63/143 (43%), Gaps = 7/143 (4%)
Query: 331 LKKIEEAEAVFEMMSNKWKLTA----RNYESLLKIYIRHKMLNKGKDLIKTMGDSGCTIG 386
LK E E + KL A +Y +LLK+Y++++ D + M ++
Sbjct: 16 LKLEERTEFAHRIWDTLQKLGAVYDVSHYNALLKVYLQNEYKFSPTDFLAKMEEANIQPN 75
Query: 387 PTTWDALVSLYVQAGEVEKADTVLQKALQQNKMKPMF-TTFMTIMEQYAKRGDVHNAEKI 445
T+ L++ Y G++E A +L + K P+ F ++ +A+ GD+ NAE I
Sbjct: 76 RVTYQRLIASYCNVGDIEGASKIL--GFMKTKDLPVTEAVFSALVTGHARAGDMENAENI 133
Query: 446 FYRLRQANYISRISPFHALAQAY 468
+R A + AL AY
Sbjct: 134 LTVMRDAGIEPGPDTYLALLNAY 156
Score = 39.7 bits (91), Expect = 0.018
Identities = 26/123 (21%), Positives = 62/123 (50%), Gaps = 2/123 (1%)
Query: 325 IEAWGRLKKIEEAEAVFEMMSNK-WKLTARNYESLLKIYIRHKMLNKGKDLIKTMGDSGC 383
I ++ + IE A + M K +T + +L+ + R + ++++ M D+G
Sbjct: 83 IASYCNVGDIEGASKILGFMKTKDLPVTEAVFSALVTGHARAGDMENAENILTVMRDAGI 142
Query: 384 TIGPTTWDALVSLYVQAGEVEKADTVLQKALQQNKMKPMFTTFMTIMEQYAKRGDVHNAE 443
GP T+ AL++ Y + G+++ L+K +++ ++ M + I+ ++K G + ++
Sbjct: 143 EPGPDTYLALLNAYAEKGDIDHVKQTLEK-VEKFELHLMDRDLLQIIFSFSKAGYLSMSQ 201
Query: 444 KIF 446
K +
Sbjct: 202 KFW 204
Score = 38.1 bits (87), Expect = 0.052
Identities = 33/171 (19%), Positives = 73/171 (42%), Gaps = 13/171 (7%)
Query: 289 CPTLLRLYAILGRADEVERIWKVCESKPRVEDCLAAIEAWGRLKKIE-EAEAVFEMMSNK 347
C +LL + R + RIW + V D + + L K+ + E F
Sbjct: 9 CGSLLPELKLEERTEFAHRIWDTLQKLGAVYD----VSHYNALLKVYLQNEYKFSPTDFL 64
Query: 348 WKLTARN-------YESLLKIYIRHKMLNKGKDLIKTMGDSGCTIGPTTWDALVSLYVQA 400
K+ N Y+ L+ Y + ++ M + + ALV+ + +A
Sbjct: 65 AKMEEANIQPNRVTYQRLIASYCNVGDIEGASKILGFMKTKDLPVTEAVFSALVTGHARA 124
Query: 401 GEVEKADTVLQKALQQNKMKPMFTTFMTIMEQYAKRGDVHNAEKIFYRLRQ 451
G++E A+ +L ++ ++P T++ ++ YA++GD+ + ++ ++ +
Sbjct: 125 GDMENAENILT-VMRDAGIEPGPDTYLALLNAYAEKGDIDHVKQTLEKVEK 174
Score = 35.4 bits (80), Expect = 0.33
Identities = 25/106 (23%), Positives = 49/106 (45%), Gaps = 3/106 (2%)
Query: 150 YRTLLANCASLENLRKTEETFNKMRELGFPVTAFACNQLLLIYKKI-DKKKIADVLLMME 208
Y+ L+A+ ++ ++ + M+ PVT + L+ + + D + ++L +M
Sbjct: 79 YQRLIASYCNVGDIEGASKILGFMKTKDLPVTEAVFSALVTGHARAGDMENAENILTVMR 138
Query: 209 KENVKPSSYTYKILIDVKGLSNDIDGMSQIVETMKAEGCELDHLTR 254
++P TY L++ DID + Q +E K E EL + R
Sbjct: 139 DAGIEPGPDTYLALLNAYAEKGDIDHVKQTLE--KVEKFELHLMDR 182
>PT09_YEAST (P32522) PET309 protein, mitochondrial precursor
Length = 965
Score = 38.9 bits (89), Expect = 0.030
Identities = 59/299 (19%), Positives = 116/299 (38%), Gaps = 47/299 (15%)
Query: 150 YRTLLANCASLENLRKTEETFNKMRELGFPVTAFACNQLLLIYKKI-DKKKIADVLLMME 208
Y L+ A + L + + ++ G T LL + K+ D + +
Sbjct: 349 YGILMYTMARIGELDSVNKLYTQLLRRGMIPTYAVLQSLLYAHYKVGDFAACFSHFELFK 408
Query: 209 KENVKPSSYTYKILIDVKGLSNDIDGMSQIVETMKAEGCELDHLTRASLARHYAAAGLTE 268
K ++ PS+ T+ I++ V ND+DG +I++ + + + +TE
Sbjct: 409 KYDITPSTATHTIMLKVYRGLNDLDGAFRILKRLSED----------------PSVEITE 452
Query: 269 KTEAILKEIEGENLKENMWVCPTLLRLYAILGRADEVERIWKVCESKPRVE----DCLAA 324
A+L ++ C T L A + ++ + ++ A
Sbjct: 453 GHFALLIQM----------CCKTTNHLIA--------QELFNLMTEHYNIQHTGKSISAL 494
Query: 325 IEAWGRLKKIEEAEAVFEMMSNKWKLTARN-----YESLLKIYIRHKMLNKGKDLIKTMG 379
++ + + EA A+FE S L+ R+ Y +K YI + NK ++L +
Sbjct: 495 MDVYIESNRPTEAIALFEKHSKN--LSWRDGLISVYNKAIKAYIGLRNANKCEELFDKIT 552
Query: 380 DSGCTIGPTTWDALVSLYVQAGE-VEKADTVLQKALQQNKMKPMFTTFMTIMEQYAKRG 437
S + + ++ V E E A +++ + ++ + +K T F IME Y K G
Sbjct: 553 TSKLAVNSEFYKMMIKFLVTLNEDCETALSIIDQLIKHSVIKVDATHFEIIMEAYDKEG 611
>Y798_METJA (Q58208) Hypothetical protein MJ0798
Length = 334
Score = 38.1 bits (87), Expect = 0.052
Identities = 57/280 (20%), Positives = 109/280 (38%), Gaps = 34/280 (12%)
Query: 186 NQLLLIYKKIDKK--KIADVLLMMEKE--------NVKPSSYTYKILIDVKGLSNDIDGM 235
N+L +Y+ IDK +I +L + K+ N+ +Y +K + N +
Sbjct: 72 NKLSTLYEDIDKLLGEIECILSLSNKDIKNWKLWKNLGDKAYLWKAYYEALFCYNKALEL 131
Query: 236 SQIVETMKAEGCELDHLTRASLARHYAAAGLTEKTEAILKEIEGENLKENMWVCPTLLRL 295
+Q E + +G L L + LA Y +EK K + G
Sbjct: 132 NQNTELLCKKGYALLKLYKRDLAIKYFEKA-SEKDRNNYKALFGLGKS-----------Y 179
Query: 296 YAILGRADEVERIWKVCESKPRVEDCLAAIEAWGRLKKIEEAEAVFEMMSNKWKLTARNY 355
Y + + ++ KV E P + + A+E G L E+ E +L +
Sbjct: 180 YLMSDNKNSIKYFEKVLELNP---NDVEALEYLGELYYEEDCEKAINYFKKALELKPDDI 236
Query: 356 ESLLKIYIRHKMLNKGKDLIKTMGDSGCTIGPTT------WDALVSLYVQAGEVEKADTV 409
+ +LK+ + L K K +K + + P ++++ +Y+ GE EKA
Sbjct: 237 DLILKVAFTYFKLKKYKHALKYF-EKALKLNPNVFELEQIYESMGRIYIYLGEDEKAIEC 295
Query: 410 LQKALQQNKMKPMFTTFMTIMEQYAKRGDVHNAEKIFYRL 449
+K + N + + I Y + G++ A++ + +L
Sbjct: 296 FEKLKEINLYH--YEIYEIIALTYEEVGNIEKAKEFYKKL 333
>RA50_SULTO (Q96YR5) DNA double-strand break repair rad50 ATPase
Length = 879
Score = 35.4 bits (80), Expect = 0.33
Identities = 58/235 (24%), Positives = 97/235 (40%), Gaps = 32/235 (13%)
Query: 120 KLDLIAKLRGLPKAEKYLEHVPNSFRGELLYRTLLANCASLENLRKTEETFNKMRELGFP 179
K+D I KLR K + N+ EL N + + ++K +E N REL
Sbjct: 158 KIDKIEKLRDSNGPIKEVMDKINNKIIELQSLEKYKNESENQKIQKEKELENIKREL--- 214
Query: 180 VTAFACNQLLLIYKKIDKKKIADVLLMMEKENVKPSSYTYKILIDVKGLSNDIDGMSQIV 239
+ L I ++ ++KK D++ + E+E K Y I + + L +DI + + V
Sbjct: 215 -------EDLNIKEEKERKKYEDIVKLNEEEEKKEKRYVELISL-LNKLKDDISELREEV 266
Query: 240 ETMKAEGCELDHLTRASLARHYAAAGLTEKTEAILK-----EIEGENLKENMWVCPTLLR 294
+ E + L + L + L E+ E I++ ++ E K + L
Sbjct: 267 KDENRLREEKEKLEKDILEKD----KLIEEKEKIIEAQNKIKLAQEKEKSLKTIKINLTD 322
Query: 295 LYAILGRADEVERIWKVCESKPRVEDCLAAIEAWGRLKKIEEAEAVFEMMSNKWK 349
L L R E+E ED IE G L+++EE E F +S++ K
Sbjct: 323 LEEKLKRKRELE------------EDYKKYIEIKGELEELEEKERKFNSLSDRLK 365
>PRSA_STAEP (Q8CNR4) Foldase protein prsA precursor (EC 5.2.1.8)
Length = 325
Score = 35.0 bits (79), Expect = 0.44
Identities = 31/112 (27%), Positives = 44/112 (38%), Gaps = 18/112 (16%)
Query: 190 LIYKKIDKKKIADVLLMMEKENVKPSSYT----------YKILIDVKGLSNDI------- 232
LI K K+ADV+ M KE + +S++ YK +D K + DI
Sbjct: 33 LISSKAGDVKVADVMKKMGKEQIANTSFSIVLNKVLADKYKDKVDTKDIDKDIKKEEKQY 92
Query: 233 DGMSQIVETMKAEGCELDHLTRASLARHYAAAGLTEKTEAILKEIEGENLKE 284
G Q +K +G D Y L +K KEI+ EN K+
Sbjct: 93 GGKDQFESMLKQQGMSFDDYKEQKKLSAYQKQLLLDKVNVSDKEIK-ENSKK 143
>ITN1_XENLA (O42287) Intersectin 1
Length = 1270
Score = 35.0 bits (79), Expect = 0.44
Identities = 48/196 (24%), Positives = 87/196 (43%), Gaps = 14/196 (7%)
Query: 66 LDKWVEKGKELSRQEIGLALNSLRRRKMYGRALQ---ALDWLESNKKLEFTEKEYASKLD 122
L+K +EK +EL RQ + RR+ R L+ L+W E N++ E + + D
Sbjct: 407 LEKQLEKQRELERQREEERRKEIERREAAKRELERQRQLEW-ERNRRQELLNQRNREQED 465
Query: 123 LIAKLRGLPKAEKYLEHVPNSFRGELLYRTLLANCASLENLRKTEETFNKMRELGFPVTA 182
++ L+ K ++ N + +L + C L R E+ NK REL
Sbjct: 466 IVV-LKAKKKTLEFELEALNDKKHQLEGKLQDIRC-RLTTQRHEIESTNKSRELRIAEIT 523
Query: 183 FACNQL----LLIYKKI-DKKKIADVLLMMEKENV-KPSSYTYKILIDVK--GLSNDIDG 234
QL L+ K I +K+ + D L +++ ++ + S T K ++ K G D
Sbjct: 524 HLQQQLQESQQLLGKMIPEKQSLIDQLKQVQQNSLHRDSLLTLKRALETKEIGRQQLRDQ 583
Query: 235 MSQIVETMKAEGCELD 250
+ ++ + +A+ E+D
Sbjct: 584 LDEVEKETRAKLQEID 599
>ITN1_HUMAN (Q15811) Intersectin 1 (SH3 domain-containing protein
1A) (SH3P17)
Length = 1721
Score = 34.7 bits (78), Expect = 0.57
Identities = 46/196 (23%), Positives = 87/196 (43%), Gaps = 14/196 (7%)
Query: 66 LDKWVEKGKELSRQEIGLALNSLRRRKMYGRALQ---ALDWLESNKKLEFTEKEYASKLD 122
L+K +EK +EL RQ + RR+ R L+ L+W E N++ E + + D
Sbjct: 409 LEKQLEKQRELERQREEERRKEIERREAAKRELERQRQLEW-ERNRRQELLNQRNKEQED 467
Query: 123 LIAKLRGLPKAEKYLEHVPNSFRGELLYRTLLANCASLENLRKTEETFNKMRELGFPVTA 182
++ L+ K ++ N + +L + C L R+ E+ NK REL
Sbjct: 468 IVV-LKAKKKTLEFELEALNDKKHQLEGKLQDIRC-RLTTQRQEIESTNKSRELRIAEIT 525
Query: 183 FACNQL----LLIYKKIDKKKIADVLLMMEKENV--KPSSYTYKILIDVKGLSND--IDG 234
QL ++ + I +K+I + L ++N + S T K ++ K L+ D
Sbjct: 526 HLQQQLQESQQMLGRLIPEKQILNDQLKQVQQNSLHRDSLVTLKRALEAKELARQHLRDQ 585
Query: 235 MSQIVETMKAEGCELD 250
+ ++ + +++ E+D
Sbjct: 586 LDEVEKETRSKLQEID 601
>T2B1_HERAU (P25257) Type II restriction enzyme HgiBI (EC 3.1.21.4)
(Endonuclease HgiBI) (R.HgiBI)
Length = 274
Score = 33.9 bits (76), Expect = 0.97
Identities = 30/131 (22%), Positives = 59/131 (44%), Gaps = 14/131 (10%)
Query: 292 LLRLYAILGRADEVERIWKVCESKPRVEDCLAAIEAWGRLKKIEEAEAV-----FEMMSN 346
++R + R D E +++ KP CL ++ R+ I++ AV + M N
Sbjct: 151 IVRADLYIQRHDGSELFFEIKSPKPSKGQCLEVMQRLLRIYTIKQQSAVPVKAFYAMAYN 210
Query: 347 KWKLTARNYESLL--KIYIRHKMLNKGKDLIKTMGDSGCTIGPTTWDALVSLYVQAGEVE 404
W ++ +Y S + K + G++ +G+ P+T+ L+ +Y + G +
Sbjct: 211 PWGISRASYRSSITKKYTDFSNAVVIGQEFWSLIGE------PSTYTELLEIYHEVGLAK 264
Query: 405 KADTVLQKALQ 415
A+ + QK LQ
Sbjct: 265 SAE-ITQKLLQ 274
>GPX4_SPIOL (O23814) Probable phospholipid hydroperoxide glutathione
peroxidase (EC 1.11.1.9) (PHGPx)
Length = 171
Score = 33.9 bits (76), Expect = 0.97
Identities = 18/46 (39%), Positives = 26/46 (56%), Gaps = 2/46 (4%)
Query: 144 FRGELLYRTLLANCASLENLRKTE--ETFNKMRELGFPVTAFACNQ 187
++G++L +A+ L N TE E + K RELG + AF CNQ
Sbjct: 30 YKGKVLLIVNVASQCGLTNSNYTEMTELYEKYRELGLEILAFPCNQ 75
>ENGA_BORPE (Q7VWL4) GTP-binding protein engA
Length = 451
Score = 33.9 bits (76), Expect = 0.97
Identities = 33/131 (25%), Positives = 59/131 (44%), Gaps = 12/131 (9%)
Query: 4 EDRLTEPDADGDSRDTAEIDINELELSDTETDSSDKKSFSSRRRSELFKAIVSVSGLSVD 63
E+R+ D G +RD EID T D++ R+R ++F+AI S +
Sbjct: 208 EERVIAFDMPGTTRDAIEIDFERDGRKYTLIDTA-----GLRKRGKVFEAIEKFSVIKTL 262
Query: 64 SALDK------WVEKGKELSRQEIGLALNSLRRRKMYGRALQALDWLESNKKLEFTEKEY 117
A++ ++ E+S Q+ +A L + A+ D L+S+++ E E+E+
Sbjct: 263 QAIEASNVVLLMIDAQAEVSEQDAHIAGFVLETGRAVVVAINKWDGLDSDQR-ERIEREF 321
Query: 118 ASKLDLIAKLR 128
KL + R
Sbjct: 322 QRKLRFLGFAR 332
>ENGA_BORPA (Q7W6Q0) GTP-binding protein engA
Length = 451
Score = 33.9 bits (76), Expect = 0.97
Identities = 33/131 (25%), Positives = 59/131 (44%), Gaps = 12/131 (9%)
Query: 4 EDRLTEPDADGDSRDTAEIDINELELSDTETDSSDKKSFSSRRRSELFKAIVSVSGLSVD 63
E+R+ D G +RD EID T D++ R+R ++F+AI S +
Sbjct: 208 EERVIAFDMPGTTRDAIEIDFERDGRKYTLIDTA-----GLRKRGKVFEAIEKFSVIKTL 262
Query: 64 SALDK------WVEKGKELSRQEIGLALNSLRRRKMYGRALQALDWLESNKKLEFTEKEY 117
A++ ++ E+S Q+ +A L + A+ D L+S+++ E E+E+
Sbjct: 263 QAIEASNVVLLMIDAQAEVSEQDAHIAGFVLETGRAVVVAINKWDGLDSDQR-ERIEREF 321
Query: 118 ASKLDLIAKLR 128
KL + R
Sbjct: 322 QRKLRFLGFAR 332
>ENGA_BORBR (Q7WHN4) GTP-binding protein engA
Length = 451
Score = 33.9 bits (76), Expect = 0.97
Identities = 33/131 (25%), Positives = 59/131 (44%), Gaps = 12/131 (9%)
Query: 4 EDRLTEPDADGDSRDTAEIDINELELSDTETDSSDKKSFSSRRRSELFKAIVSVSGLSVD 63
E+R+ D G +RD EID T D++ R+R ++F+AI S +
Sbjct: 208 EERVIAFDMPGTTRDAIEIDFERDGRKYTLIDTA-----GLRKRGKVFEAIEKFSVIKTL 262
Query: 64 SALDK------WVEKGKELSRQEIGLALNSLRRRKMYGRALQALDWLESNKKLEFTEKEY 117
A++ ++ E+S Q+ +A L + A+ D L+S+++ E E+E+
Sbjct: 263 QAIEASNVVLLMIDAQAEVSEQDAHIAGFVLETGRAVVVAINKWDGLDSDQR-ERIEREF 321
Query: 118 ASKLDLIAKLR 128
KL + R
Sbjct: 322 QRKLRFLGFAR 332
>CE29_HUMAN (O15078) Centrosomal protein Cep290
Length = 1539
Score = 33.9 bits (76), Expect = 0.97
Identities = 52/259 (20%), Positives = 104/259 (40%), Gaps = 28/259 (10%)
Query: 2 EIEDRLTEPDADGDSRDTAEIDINELELSDTETDSSDKKSFSSRRRSELFKAIVSVSGLS 61
E+ D +++ +D D + E++ NE+EL E + S +RR+ E+ A
Sbjct: 181 ELADSVSKAVSDADRQRILELEKNEMELK-VEVSKLREISDIARRQVEILNAQQQSRDKE 239
Query: 62 VDSALDKWVEKGKELSRQEIGLALNSLRRRKMYGRALQALDWLESNKKLEFTEKEYASKL 121
V+S + ++ + + + L+ + N L+ +E KL
Sbjct: 240 VESLRMQLLDYQAQSDEKSLIAKLH------------------QHNVSLQLSEATALGKL 281
Query: 122 DLI-AKLRGLPKAEKYLEHVPNSFRGELLYRTLLANCASLENLRKTEETFNKMRELGFPV 180
+ I +KL+ + LE + + + LY L ++LR+T ++ + P+
Sbjct: 282 ESITSKLQKMEAYNLRLEQKLDE-KEQALYYARLEGRNRAKHLRQTIQSLRRQFSGALPL 340
Query: 181 TAFACNQLLLIYKKIDKKKIADVLLMMEKENVKPSSYTYKILIDVKGLS------NDIDG 234
+I + DK KI + ++E+ + T ++ + +KGL D G
Sbjct: 341 AQQEKFSKTMIQLQNDKLKIMQEMKNSQQEHRNMENKTLEMELKLKGLEELISTLKDTKG 400
Query: 235 MSQIVE-TMKAEGCELDHL 252
+++ MK E L L
Sbjct: 401 AQKVINWHMKIEELRLQEL 419
>YA88_AQUAE (O67178) Hypothetical protein AQ_1088
Length = 761
Score = 33.5 bits (75), Expect = 1.3
Identities = 30/120 (25%), Positives = 52/120 (43%), Gaps = 12/120 (10%)
Query: 257 LARHYAAAGLTEKTEAILKEIEGENLKENMWVCPTLLRLYAILGRADEVERIWKVCESKP 316
L ++ +G +K E +L ++ +L + L LY LGR ++ ER+ K
Sbjct: 41 LGIYHFLSGEPQKAEELLSQVSENSLNSAQGLSDLGL-LYFFLGRVEDAERVLKKALKFS 99
Query: 317 RVEDCLAAIEAWGRLKKIEEAEAVFEMMSNKWK----LTARNYESLLKIYIRHKMLNKGK 372
V+D L + RL + ++ E + W+ L E L + + H LNKG+
Sbjct: 100 DVDDAL-----YARLGALYYSQGKLEEAQHYWERALSLNPNKVEILYNLGVLH--LNKGE 152
>RA50_THEMA (Q9X1X1) Probable DNA double-strand break repair rad50
ATPase
Length = 852
Score = 33.5 bits (75), Expect = 1.3
Identities = 55/276 (19%), Positives = 111/276 (39%), Gaps = 38/276 (13%)
Query: 18 DTAEIDINELELSDTETDSSDKKSFSSRRRSELFKAIVSVSGLSVDSALDKWVEKGKELS 77
+ E +I+E E D + + + R + +++ + ++ +K +
Sbjct: 447 EAVEFNIDEFEKLDQKRSELENTLNVLKERKKSLSSLIEDLLMKIEEG-----KKNLKSI 501
Query: 78 RQEIGLALNSLRRRKMYGRALQALDWLESNKKLEFTEKEYASKLDLIAKLRGLPKAEKYL 137
R +I L R + LD E KKL E+E S + K+ +
Sbjct: 502 RNQIEKIEEELHRLGYSEDLEEKLD--EKRKKLRKIEEERHS---ISQKITAADVQISQI 556
Query: 138 EHVPNSFRGELLYR--TLLANCASLENLRKTEETFNKMRELGFPVTAFACNQLLLIYKKI 195
E+ +GE+ + TL ++ L+ + F+++R++G F ++L+ +
Sbjct: 557 ENQLKEIKGEIEAKRETLKEQREEMDQLKS--DFFDRLRKIGIGFEEF---RILVKEEVK 611
Query: 196 DKKK-----------IADVLLMMEKENVKPSSYTYKILIDVKGLSNDIDGMSQIVETMKA 244
D +K + + L +E ENV+ S Y+ + N ++ +SQ + ++
Sbjct: 612 DAEKELGVVETEIRLLEESLKELESENVRDVSEDYE------KVRNQLEALSQEISDLER 665
Query: 245 EGCELDHLTRASLARHYAAAGLTEKTEAILKEIEGE 280
+ L+HL +L R L +K LKE+ E
Sbjct: 666 KEGRLNHLIEETLRRERELKSLEKK----LKEMSDE 697
>T2E1_HERAU (P25260) Type II restriction enzyme HgiEI (EC 3.1.21.4)
(Endonuclease HgiEI) (R.HgiEI)
Length = 274
Score = 33.1 bits (74), Expect = 1.7
Identities = 30/131 (22%), Positives = 58/131 (43%), Gaps = 14/131 (10%)
Query: 292 LLRLYAILGRADEVERIWKVCESKPRVEDCLAAIEAWGRLKKIEEAEAV-----FEMMSN 346
++R + R D E +++ KP CL ++ R+ I++ AV + M N
Sbjct: 151 IVRADLYIQRHDGSELFFEIKSPKPNKGQCLEVMQRLLRIYTIKQQSAVPVKAFYAMAYN 210
Query: 347 KWKLTARNYES--LLKIYIRHKMLNKGKDLIKTMGDSGCTIGPTTWDALVSLYVQAGEVE 404
W ++ +Y S K + G++ +G+ P+T+ L+ +Y + G +
Sbjct: 211 PWGISRASYRSSNTKKYTDFSNAVVIGQEFWSLIGE------PSTYTELLEIYHEVGLAK 264
Query: 405 KADTVLQKALQ 415
A+ + QK LQ
Sbjct: 265 SAE-ITQKLLQ 274
>DPO1_BACST (P52026) DNA polymerase I (EC 2.7.7.7) (POL I)
Length = 876
Score = 33.1 bits (74), Expect = 1.7
Identities = 29/97 (29%), Positives = 45/97 (45%), Gaps = 18/97 (18%)
Query: 194 KIDKKKIADVLLMMEKENVKPSSYTYKILIDVKGL----SNDIDGMSQIVETMKAEGCEL 249
+I KK I D+ + V+ T + ++D+KGL S++I G+ I E
Sbjct: 148 EITKKGITDIESYTPETVVEKYGLTPEQIVDLKGLMGDKSDNIPGVPGIGEK-------- 199
Query: 250 DHLTRASLARHYAAAGLTEKTEAILKEIEGENLKENM 286
T L + + G E A + EI+GE LKEN+
Sbjct: 200 ---TAVKLLKQF---GTVENVLASIDEIKGEKLKENL 230
>YO91_CAEEL (P41842) Hypothetical protein T20B12.1 in chromosome III
Length = 787
Score = 32.7 bits (73), Expect = 2.2
Identities = 10/38 (26%), Positives = 24/38 (62%)
Query: 390 WDALVSLYVQAGEVEKADTVLQKALQQNKMKPMFTTFM 427
WD ++ Y Q G+++KA+T++++ ++Q M ++
Sbjct: 408 WDGVIDCYKQLGQMDKAETLIRRLIEQKPNDSMLHVYL 445
>HISX_PROMP (Q7V004) Histidinol dehydrogenase (EC 1.1.1.23) (HDH)
Length = 428
Score = 32.7 bits (73), Expect = 2.2
Identities = 15/44 (34%), Positives = 27/44 (61%), Gaps = 1/44 (2%)
Query: 301 RADEV-ERIWKVCESKPRVEDCLAAIEAWGRLKKIEEAEAVFEM 343
+A EV + ++K+ E+ PR E C+ +I+ WG + E E+ E+
Sbjct: 275 QAQEVFDEVFKIIENHPRKEICIQSIKNWGLIAICENLESCVEL 318
>APN1_YEAST (P22936) DNA-(apurinic or apyrimidinic site) lyase 1 (EC
4.2.99.18) (AP endonuclease 1) (Apurinic-apyrimidinic
endonuclease 1)
Length = 366
Score = 32.7 bits (73), Expect = 2.2
Identities = 16/43 (37%), Positives = 24/43 (55%), Gaps = 1/43 (2%)
Query: 94 YGRALQALDWLESNKKLEFTE-KEYASKLDLIAKLRGLPKAEK 135
YG ++ ++WLES + E E KEY K D + KL + E+
Sbjct: 282 YGNEIKLMEWLESKSESELLEDKEYKEKNDTLQKLGAKSRKEQ 324
Database: sprot
Posted date: Nov 25, 2004 10:54 AM
Number of letters in database: 59,974,054
Number of sequences in database: 164,201
Lambda K H
0.317 0.133 0.373
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 53,004,078
Number of Sequences: 164201
Number of extensions: 2125333
Number of successful extensions: 7412
Number of sequences better than 10.0: 31
Number of HSP's better than 10.0 without gapping: 3
Number of HSP's successfully gapped in prelim test: 28
Number of HSP's that attempted gapping in prelim test: 7387
Number of HSP's gapped (non-prelim): 54
length of query: 488
length of database: 59,974,054
effective HSP length: 114
effective length of query: 374
effective length of database: 41,255,140
effective search space: 15429422360
effective search space used: 15429422360
T: 11
A: 40
X1: 16 ( 7.3 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 41 (21.6 bits)
S2: 68 (30.8 bits)
Medicago: description of AC135463.1