
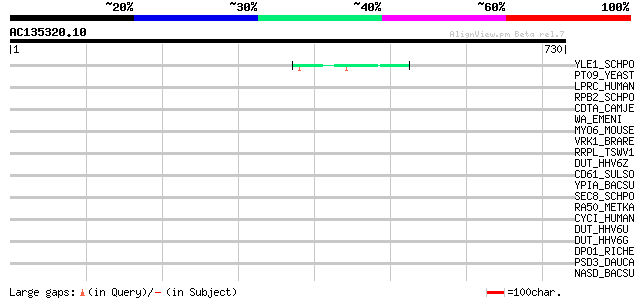
BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query= AC135320.10 - phase: 0
(730 letters)
Database: sprot
164,201 sequences; 59,974,054 total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
YLE1_SCHPO (Q10451) Hypothetical protein C1093.01 in chromosome I 49 4e-05
PT09_YEAST (P32522) PET309 protein, mitochondrial precursor 43 0.003
LPRC_HUMAN (P42704) 130 kDa leucine-rich protein (LRP 130) (GP13... 42 0.006
RPB2_SCHPO (Q02061) DNA-directed RNA polymerase II 138 kDa polyp... 34 1.2
CDTA_CAMJE (Q46100) Cytolethal distending toxin subunit A precur... 34 1.2
WA_EMENI (Q03149) Conidial yellow pigment biosynthesis polyketid... 34 1.6
MYO6_MOUSE (Q64331) Myosin VI 34 1.6
VRK1_BRARE (Q7ZUS1) Serine/threonine-protein kinase VRK1 (EC 2.7... 33 2.7
RRPL_TSWV1 (P28976) RNA-directed RNA polymerase (EC 2.7.7.48) (L... 33 3.5
DUT_HHV6Z (P52541) Deoxyuridine 5'-triphosphate nucleotidohydrol... 33 3.5
CD61_SULSO (Q980N4) Cell division control protein 6 homolog 1 (C... 33 3.5
YPIA_BACSU (P54389) Hypothetical protein ypiA 32 4.6
SEC8_SCHPO (O74562) Exocyst complex component sec8 32 4.6
RA50_METKA (Q8TXI4) DNA double-strand break repair rad50 ATPase 32 4.6
CYCI_HUMAN (Q14094) Cyclin I 32 4.6
DUT_HHV6U (Q06095) Deoxyuridine 5'-triphosphate nucleotidohydrol... 32 6.0
DUT_HHV6G (P30007) Deoxyuridine 5'-triphosphate nucleotidohydrol... 32 6.0
DPO1_RICHE (Q9RLB6) DNA polymerase I (EC 2.7.7.7) (POL I) 32 6.0
PSD3_DAUCA (Q06364) Probable 26S proteasome non-ATPase regulator... 32 7.8
NASD_BACSU (P42435) Nitrite reductase [NAD(P)H] (EC 1.7.1.4) 32 7.8
>YLE1_SCHPO (Q10451) Hypothetical protein C1093.01 in chromosome I
Length = 1261
Score = 49.3 bits (116), Expect = 4e-05
Identities = 42/162 (25%), Positives = 66/162 (39%), Gaps = 21/162 (12%)
Query: 373 ALAQANW-----IHTYVDRSGFGRALSVNNALIDMYAKCGNLVKAREVFENMPRKNVISW 427
+L +ANW + + S F R +N D G + +A ++
Sbjct: 844 SLEKANWFMALILDAMILSSSFARQFKSSNLFCDNMKMLGYIPRAS------------TF 891
Query: 428 SSMINAFAMHGNAD---SAIKLFRRMKEVNIEPNGVTFIGVLYACGHAGLVEEGEKLFSS 484
+ +IN G+ D +A+ +F K N++P+ + VL G A E KLF
Sbjct: 892 AHLINNSTRRGDTDDATTALNIFEETKRHNVKPSVFLYNAVLSKLGRARRTTECWKLFQE 951
Query: 485 MINEHGISPTREHYGCMVDLYCRANFLRKAIELIETMPFAPN 526
M E G+ PT YG +++ CR A +L M PN
Sbjct: 952 M-KESGLLPTSVTYGTVINAACRIGDESLAEKLFAEMENQPN 992
Score = 42.4 bits (98), Expect = 0.004
Identities = 29/106 (27%), Positives = 45/106 (42%), Gaps = 4/106 (3%)
Query: 412 AREVFENMPRKNV----ISWSSMINAFAMHGNADSAIKLFRRMKEVNIEPNGVTFIGVLY 467
A +FE R NV ++++++ KLF+ MKE + P VT+ V+
Sbjct: 910 ALNIFEETKRHNVKPSVFLYNAVLSKLGRARRTTECWKLFQEMKESGLLPTSVTYGTVIN 969
Query: 468 ACGHAGLVEEGEKLFSSMINEHGISPTREHYGCMVDLYCRANFLRK 513
A G EKLF+ M N+ P Y M+ + F R+
Sbjct: 970 AACRIGDESLAEKLFAEMENQPNYQPRVAPYNTMIQFEVQTMFNRE 1015
Score = 42.0 bits (97), Expect = 0.006
Identities = 91/463 (19%), Positives = 175/463 (37%), Gaps = 60/463 (12%)
Query: 93 NQLLRHLSRSSFPEKT-----IFLYHNLRAINAFALDRFSFPSLLKAVSKVSAFN----- 142
N L+ + S+ PE+ F + L N F L +P+L+ +SK F+
Sbjct: 771 NSLVSKMLTSASPEQVDVNILFFQFGKLIETNKF-LHPEVYPTLISVLSKNKRFDAVQRV 829
Query: 143 --HGLEIHG-LASKLGFVDDPFIQTGLIAMYAS---CRRIMDARLLFDKMCH----PDAV 192
H ++ +++K + F+ L AM S R+ + L D M P A
Sbjct: 830 FEHSKHLYRKISTKSLEKANWFMALILDAMILSSSFARQFKSSNLFCDNMKMLGYIPRAS 889
Query: 193 AWNMIIDGYCQNGHYDDA---LRLFEDMRSSDMKPDSVILCTVLSACGHAGNLSYGRTIH 249
+ +I+ + G DDA L +FE+ + ++KP + VLS G A + +
Sbjct: 890 TFAHLINNSTRRGDTDDATTALNIFEETKRHNVKPSVFLYNAVLSKLGRARRTTECWKLF 949
Query: 250 EFVKDNGYAIDSHLQTALINMYANCGAMDLARKIYDGLSSK-----HLIVSTAMLSGYAK 304
+ +K++G S +IN G LA K++ + ++ + M+ +
Sbjct: 950 QEMKESGLLPTSVTYGTVINAACRIGDESLAEKLFAEMENQPNYQPRVAPYNTMIQFEVQ 1009
Query: 305 LGMVKD-ARFIFDQMIERDLV----CWSAMISGYAESDQPQ-EALKLFDEMLQKRSVPDQ 358
++ A F ++++ D+ + ++ Y ++K E++++ VP
Sbjct: 1010 TMFNREKALFYYNRLCATDIEPSSHTYKLLMDAYGTLKPVNVGSVKAVLELMERTDVP-- 1067
Query: 359 ITMLSVISACSHVGALAQANWIHTYVDRSGFGRALSVNNALIDMYAKCGNLVKAREVFEN 418
+ ++ A +IH G +S A Y N + + E
Sbjct: 1068 ------------ILSMHYAAYIHI------LGNVVSDVQAATSCYM---NALAKHDAGEI 1106
Query: 419 MPRKNVISWSSMINAFAMHGNADSAIKLFRRMKEVNIEPNGVTFIGVLYACGHAGLVEEG 478
N+ + S I + + I++ MK N+ N ++ G++ +
Sbjct: 1107 QLDANL--FQSQIESLIANDRIVEGIQIVSDMKRYNVSLNAYIVNALIKGFTKVGMISKA 1164
Query: 479 EKLFSSMINEHGISPTREHYGCMVDLYCRANFLRKAIELIETM 521
F + E Y MV Y N RKA+E++E +
Sbjct: 1165 RYYFDLLECEGMSGKEPSTYENMVRAYLSVNDGRKAMEIVEQL 1207
Score = 35.4 bits (80), Expect = 0.54
Identities = 21/99 (21%), Positives = 50/99 (50%), Gaps = 6/99 (6%)
Query: 289 SKHLIVSTAMLSGYAKLGMVKDARFIFD-----QMIERDLVCWSAMISGYAESDQPQEAL 343
S + + A++ G+ K+GM+ AR+ FD M ++ + M+ Y + ++A+
Sbjct: 1142 SLNAYIVNALIKGFTKVGMISKARYYFDLLECEGMSGKEPSTYENMVRAYLSVNDGRKAM 1201
Query: 344 KLFDEMLQKRSVPDQITMLSVISACSHVGALAQANWIHT 382
++ +++ +KR + +S + SH+G + ++T
Sbjct: 1202 EIVEQLKRKRYPLPVVNRISSL-VNSHMGQKPKRRSLNT 1239
>PT09_YEAST (P32522) PET309 protein, mitochondrial precursor
Length = 965
Score = 43.1 bits (100), Expect = 0.003
Identities = 68/344 (19%), Positives = 137/344 (39%), Gaps = 35/344 (10%)
Query: 192 VAWNMIIDGYCQNGHYDD---ALRLFED-MRSSDMKPDSVILCTVLSACGHAGNLSYGRT 247
V W +++G+C+ +Y + LR F MR S+++PD LSY +
Sbjct: 188 VKWMKLVNGHCEFTNYMENKIVLRNFLSFMRQSNVRPDY---------------LSYLKA 232
Query: 248 IHEFVKDNGYAIDSHLQTALINMYANCGAMDLARKIYDGLSSKHL-IVS---TAMLSGYA 303
I G AI S T L+ + A +++ +L IVS T +L Y
Sbjct: 233 IQ---LTQGPAIASQFATTLLFLLTYIRKFSSAEAVWNYKCEHNLPIVSSDLTCILKTYC 289
Query: 304 ---KLGMVKDARFIFDQMIERDLVCWSAMISGYAESDQPQEALKLFDEMLQKRSVPDQIT 360
K +V + + + D + ++ ++ + F+ + +P
Sbjct: 290 HMQKFNLVSSTYWKYPDA-QHDQNQFDYLLVAHSRLHNWDALQQQFNALFGIGKLPSIQH 348
Query: 361 MLSVISACSHVGALAQANWIHTYVDRSGFGRALSVNNALIDMYAKCGNLVKAREVFENMP 420
++ + +G L N ++T + R G +V +L+ + K G+ FE
Sbjct: 349 YGILMYTMARIGELDSVNKLYTQLLRRGMIPTYAVLQSLLYAHYKVGDFAACFSHFELFK 408
Query: 421 RKNVISWSS----MINAFAMHGNADSAIKLFRRMKE-VNIEPNGVTFIGVLYACGHAGLV 475
+ ++ ++ M+ + + D A ++ +R+ E ++E F ++ C
Sbjct: 409 KYDITPSTATHTIMLKVYRGLNDLDGAFRILKRLSEDPSVEITEGHFALLIQMCCKTTNH 468
Query: 476 EEGEKLFSSMINEHGISPTREHYGCMVDLYCRANFLRKAIELIE 519
++LF+ M + I T + ++D+Y +N +AI L E
Sbjct: 469 LIAQELFNLMTEHYNIQHTGKSISALMDVYIESNRPTEAIALFE 512
Score = 40.4 bits (93), Expect = 0.017
Identities = 60/295 (20%), Positives = 115/295 (38%), Gaps = 22/295 (7%)
Query: 205 GHYDDALRLFEDMRSSDMKPDSVILCTVLSACGHAGNLSYGRTIHEFVKDNGYAIDSHLQ 264
G D +L+ + M P +L ++L A G+ + + E K +
Sbjct: 360 GELDSVNKLYTQLLRRGMIPTYAVLQSLLYAHYKVGDFAACFSHFELFKKYDITPSTATH 419
Query: 265 TALINMYANCGAMDLARKIYDGLSSKHLIVSTA-----MLSGYAKLGMVKDARFIFDQMI 319
T ++ +Y +D A +I LS + T ++ K A+ +F+ M
Sbjct: 420 TIMLKVYRGLNDLDGAFRILKRLSEDPSVEITEGHFALLIQMCCKTTNHLIAQELFNLMT 479
Query: 320 ERDLV-----CWSAMISGYAESDQPQEALKLFDEMLQKRSVPDQITMLSVISACSHVGAL 374
E + SA++ Y ES++P EA+ LF++ + S D + + + +++G L
Sbjct: 480 EHYNIQHTGKSISALMDVYIESNRPTEAIALFEKHSKNLSWRDGLISVYNKAIKAYIG-L 538
Query: 375 AQANWIHTYVDRSGFGRALSVNNALIDMYAKC-----GNLVKAREVFENMPRKNVIS--- 426
AN D+ + L+VN+ M K + A + + + + +VI
Sbjct: 539 RNANKCEELFDKITTSK-LAVNSEFYKMMIKFLVTLNEDCETALSIIDQLIKHSVIKVDA 597
Query: 427 --WSSMINAFAMHGNADSAIKLFRRMKEVNIEPNGVTFIGVLYACGHAGLVEEGE 479
+ ++ A+ G D I L++ M + + N +L A L E
Sbjct: 598 THFEIIMEAYDKEGYRDGIINLYKTMSQNKVPANSKILYYILKAVAKKSLQNNEE 652
>LPRC_HUMAN (P42704) 130 kDa leucine-rich protein (LRP 130) (GP130)
(Leucine-rich PPR-motif containing protein)
Length = 1273
Score = 42.0 bits (97), Expect = 0.006
Identities = 30/130 (23%), Positives = 51/130 (39%), Gaps = 4/130 (3%)
Query: 153 KLGFVDDPFIQTGLIAMYASCRRIMDARLLFDKM----CHPDAVAWNMIIDGYCQNGHYD 208
KLG V D L+ +Y KM P+ V + +I YC G +
Sbjct: 34 KLGAVYDVSHYNALLKVYLQNEYKFSPTDFLAKMEEANIQPNRVTYQRLIASYCNVGDIE 93
Query: 209 DALRLFEDMRSSDMKPDSVILCTVLSACGHAGNLSYGRTIHEFVKDNGYAIDSHLQTALI 268
A ++ M++ D+ + +++ AG++ I ++D G AL+
Sbjct: 94 GASKILGFMKTKDLPVTEAVFSALVTGHARAGDMENAENILTVMRDAGIEPGPDTYLALL 153
Query: 269 NMYANCGAMD 278
N YA G +D
Sbjct: 154 NAYAEKGDID 163
Score = 40.0 bits (92), Expect = 0.022
Identities = 28/176 (15%), Positives = 79/176 (43%), Gaps = 4/176 (2%)
Query: 322 DLVCWSAMISGYAESDQPQEALKLFDEMLQKRSVPDQITMLSVISACSHVGALAQANWIH 381
D+ ++A++ Y +++ +M + P+++T +I++ +VG + A+ I
Sbjct: 40 DVSHYNALLKVYLQNEYKFSPTDFLAKMEEANIQPNRVTYQRLIASYCNVGDIEGASKIL 99
Query: 382 TYVDRSGFGRALSVNNALIDMYAKCGNLVKAREVFENMPRKNV----ISWSSMINAFAMH 437
++ +V +AL+ +A+ G++ A + M + ++ +++NA+A
Sbjct: 100 GFMKTKDLPVTEAVFSALVTGHARAGDMENAENILTVMRDAGIEPGPDTYLALLNAYAEK 159
Query: 438 GNADSAIKLFRRMKEVNIEPNGVTFIGVLYACGHAGLVEEGEKLFSSMINEHGISP 493
G+ D + ++++ + + ++++ AG + +K + E P
Sbjct: 160 GDIDHVKQTLEKVEKFELHLMDRDLLQIIFSFSKAGYLSMSQKFWKKFTCERRYIP 215
Score = 36.6 bits (83), Expect = 0.24
Identities = 36/189 (19%), Positives = 83/189 (43%), Gaps = 17/189 (8%)
Query: 190 DAVAWNMIIDGYCQNGHYDDALRLFEDMRSSDMKPDSVILCTVLSACGHAGNLSYGRTIH 249
D +N ++ Y QN + M ++++P+ V ++++ + G++ I
Sbjct: 40 DVSHYNALLKVYLQNEYKFSPTDFLAKMEEANIQPNRVTYQRLIASYCNVGDIEGASKIL 99
Query: 250 EFVKDNGYAIDSHLQTALINMYANCGAMDLARKIYDGLSSKHL----IVSTAMLSGYAKL 305
F+K + + +AL+ +A G M+ A I + + A+L+ YA+
Sbjct: 100 GFMKTKDLPVTEAVFSALVTGHARAGDMENAENILTVMRDAGIEPGPDTYLALLNAYAEK 159
Query: 306 GMVKDARFIFDQ-------MIERDL--VCWSAMISGYAESDQPQEALKLFDEMLQKRSVP 356
G + + ++ +++RDL + +S +GY Q+ K F ++R +P
Sbjct: 160 GDIDHVKQTLEKVEKFELHLMDRDLLQIIFSFSKAGYL--SMSQKFWKKF--TCERRYIP 215
Query: 357 DQITMLSVI 365
D + ++ ++
Sbjct: 216 DAMNLILLL 224
Score = 35.4 bits (80), Expect = 0.54
Identities = 50/255 (19%), Positives = 101/255 (39%), Gaps = 39/255 (15%)
Query: 398 ALIDM---YAKCGNLVKAREVFENMPRKNVI---SWSSMINAFAMHGNADSAIKLFRRMK 451
ALI++ + K + + +E F+ + V+ ++ ++ A HG AIK+ + MK
Sbjct: 595 ALINLCCRHDKVEDALNLKEEFDRLDSSAVLDTGNYLGLVRVLAKHGKLQDAIKILKEMK 654
Query: 452 EVNI---EPNGVTFIGVLYACGHAGLVEEGEKLFSSMINEHGISPTREHYGCMVDLYCR- 507
E ++ + ++F +L G +E ++L +++ P+ +V ++
Sbjct: 655 EKDVLIKDTTALSFFHMLNGAALRGEIETVKQLHEAIVTLGLAEPSTNISFPLVTVHLEK 714
Query: 508 ---ANFLRKAIELIETMPFAPNVIIWGSLMSACQVHGEAELGEFAAKRLLELEPDHDGAL 564
+ L AI+ E P + ++ GE +L ++ ++ G +
Sbjct: 715 GDLSTALEVAIDCYEKYKVLPRI---HDVLCKLVEKGETDL----IQKAMDFVSQEQGEM 767
Query: 565 VVLSNIYAKEKRWNDVGLIRKSMSYKGISKEKA-----SSRIEINNQVHMFMMADRYHKQ 619
V+L +++ + + +K + GI A R NNQV
Sbjct: 768 VMLYDLFFAFLQTGNYKEAKKIIETPGIRARSARLQWFCDRCVANNQV------------ 815
Query: 620 SDEIYEKLDEVVSKL 634
E EKL E+ KL
Sbjct: 816 --ETLEKLVELTQKL 828
Score = 32.3 bits (72), Expect = 4.6
Identities = 20/99 (20%), Positives = 45/99 (45%), Gaps = 1/99 (1%)
Query: 423 NVISWSSMINAFAMHGNADSAIKLFRRMKEVNIEPNGVTFIGVLYACGHAGLVEEGEKLF 482
+V +++++ + + S +M+E NI+PN VT+ ++ + + G +E K+
Sbjct: 40 DVSHYNALLKVYLQNEYKFSPTDFLAKMEEANIQPNRVTYQRLIASYCNVGDIEGASKIL 99
Query: 483 SSMINEHGISPTREHYGCMVDLYCRANFLRKAIELIETM 521
M + T + +V + RA + A ++ M
Sbjct: 100 GFM-KTKDLPVTEAVFSALVTGHARAGDMENAENILTVM 137
>RPB2_SCHPO (Q02061) DNA-directed RNA polymerase II 138 kDa
polypeptide (EC 2.7.7.6) (RNA polymerase II subunit 2)
Length = 1210
Score = 34.3 bits (77), Expect = 1.2
Identities = 24/115 (20%), Positives = 50/115 (42%), Gaps = 8/115 (6%)
Query: 325 CWSAMISGYAESDQPQEALKLFDEMLQK--RSVPDQITMLSVISACSHVGALA------Q 376
CW+ + S + E+ ++ L FDE +Q + + D + L++ H GA +
Sbjct: 17 CWTVISSFFEETSLARQQLFSFDEFVQNTMQEIVDDDSTLTLDQYAQHTGAQGDVTRRYE 76
Query: 377 ANWIHTYVDRSGFGRALSVNNALIDMYAKCGNLVKAREVFENMPRKNVISWSSMI 431
N+ Y+ R A + A+ NL + ++ +M +K +++ S +
Sbjct: 77 INFGQIYLSRPTMTEADGSTTTMFPQEARLRNLTYSSPLYVDMRKKVMVAADSNV 131
>CDTA_CAMJE (Q46100) Cytolethal distending toxin subunit A precursor
(CDT A)
Length = 268
Score = 34.3 bits (77), Expect = 1.2
Identities = 23/64 (35%), Positives = 34/64 (52%), Gaps = 5/64 (7%)
Query: 334 AESDQPQEALKLFDEML---QKRSVPDQITMLSVISACSHVGALAQANWI--HTYVDRSG 388
A P+ + F+++L + +V D +T+L A V ALAQ NWI +T +D G
Sbjct: 88 ANGINPRFKDEAFNDVLIFENRPAVSDFLTILGPSGAALTVWALAQGNWIWGYTLIDSKG 147
Query: 389 FGRA 392
FG A
Sbjct: 148 FGDA 151
>WA_EMENI (Q03149) Conidial yellow pigment biosynthesis polyketide
synthase (EC 2.3.1.-) (PKS)
Length = 2157
Score = 33.9 bits (76), Expect = 1.6
Identities = 30/106 (28%), Positives = 47/106 (44%), Gaps = 12/106 (11%)
Query: 254 DNGYAIDSHLQTALINMYA---NCGAMDLARKIYDGLSSKHLIVSTAMLSGYAKLGMVKD 310
D G+ SH Q+ L+ + +C A+ A I + L +V A+ G V
Sbjct: 100 DLGHTFPSHSQSQLVGLCTGLLSCAAVSCASNIGELLKPAVEVVVVALRLGLC----VYR 155
Query: 311 ARFIFDQMIERDLVCWSAMISGYAESDQPQEALKLFDEMLQKRSVP 356
R +F Q L WSA++SG +ES E L D+ ++ +P
Sbjct: 156 VRKLFGQDQAAPL-SWSALVSGLSES----EGTSLIDKFTRRNVIP 196
>MYO6_MOUSE (Q64331) Myosin VI
Length = 1265
Score = 33.9 bits (76), Expect = 1.6
Identities = 27/101 (26%), Positives = 49/101 (47%), Gaps = 11/101 (10%)
Query: 546 GEFAA-KRLLELEPDHDGALVVLSNIYAKEKRWNDVGLIRKS-MSYKGISKEKASSRIEI 603
G+FA ++++ +PDH LV N++ RW V S + K K +A + I++
Sbjct: 764 GKFAEFDQIMKSDPDHLAELVKRVNLWLVCSRWKKVQWCSLSVIKLKNKIKYRAEACIKM 823
Query: 604 NNQVHMFMMADRYHKQSDEI---------YEKLDEVVSKLK 635
+ M++ R++ + D + +K +EVVS LK
Sbjct: 824 QKPIRMWLCKRRHNPRIDGLVKVGTLKKRLDKFNEVVSALK 864
>VRK1_BRARE (Q7ZUS1) Serine/threonine-protein kinase VRK1 (EC
2.7.1.37) (Vaccinia-related kinase 1)
Length = 425
Score = 33.1 bits (74), Expect = 2.7
Identities = 21/78 (26%), Positives = 38/78 (47%), Gaps = 2/78 (2%)
Query: 550 AKRLLELEPDHDGALVVLSNIYAKEKRWNDVGLIRKS--MSYKGISKEKASSRIEINNQV 607
A ++++EP +G L Y + + + +G KS M Y G+ K S E N +
Sbjct: 67 APYVIKVEPSDNGPLFSELKFYMRAAKPDLIGAWMKSKKMEYLGVPKYWGSGFHEKNGKR 126
Query: 608 HMFMMADRYHKQSDEIYE 625
+ FM+ DR+ +++E
Sbjct: 127 YRFMVMDRFGTDLQKLFE 144
>RRPL_TSWV1 (P28976) RNA-directed RNA polymerase (EC 2.7.7.48) (L
protein)
Length = 2875
Score = 32.7 bits (73), Expect = 3.5
Identities = 34/144 (23%), Positives = 63/144 (43%), Gaps = 9/144 (6%)
Query: 566 VLSNIYAKEKRWNDVGLIRKSMSYKGISKEKASSRIEINNQVHMFMMADRYHKQSDEIYE 625
+ +IY K+ ++D L S G KA S + N ++ +M + D +
Sbjct: 1035 IFEDIYPKKAMFDDKDLF----SINGALNVKALSDYYLGNIENVGLMRSEIENKEDFLSP 1090
Query: 626 --KLDEVVSKLKLVGYKP-STSGILIDLEEEDKKELVLWHSEKLAVCYGLISRRNESCIR 682
K+ + S K ST I+ L++ +++ W LA+ GLI NE R
Sbjct: 1091 CYKISTLKSSKKCSQSNIISTDEIIECLQDAKIQDIENWKGNNLAIIKGLIRTYNEEKNR 1150
Query: 683 IVKNLRICEDCHSFMKLVSKVYQI 706
+V+ ++C + + L+ K+ +I
Sbjct: 1151 LVEFFE--DNCVNSLYLIEKLKEI 1172
>DUT_HHV6Z (P52541) Deoxyuridine 5'-triphosphate nucleotidohydrolase
(EC 3.6.1.23) (dUTPase) (dUTP pyrophosphatase)
Length = 376
Score = 32.7 bits (73), Expect = 3.5
Identities = 12/53 (22%), Positives = 26/53 (48%)
Query: 419 MPRKNVISWSSMINAFAMHGNADSAIKLFRRMKEVNIEPNGVTFIGVLYACGH 471
+P K +I +I + + + +IK+F ++ P G+ +++ CGH
Sbjct: 291 IPSKEIIKLGLLIETYIWNKDTIPSIKIFNSTRKTIYIPTGICIARIIFTCGH 343
>CD61_SULSO (Q980N4) Cell division control protein 6 homolog 1 (CDC6
homolog 1)
Length = 397
Score = 32.7 bits (73), Expect = 3.5
Identities = 48/180 (26%), Positives = 78/180 (42%), Gaps = 33/180 (18%)
Query: 551 KRLLELEPDHDGALV-VLSNIYAKEKRWNDVGLIRKSMSYKGISKEKASSRIEINNQVHM 609
+RL++ D+ +V VL I A K++ND L + S ++K K S I I N V
Sbjct: 128 RRLVKAVRDYGSQVVIVLDEIDAFVKKYNDDILYKLSRINSEVNKSKISF-IGITNDVKF 186
Query: 610 FMMADRYHKQS---DEIY------EKLDEVVSKLKLVGYKPSTSGILIDLEEEDKKELVL 660
+ D K S +EI E+L+++++K + +KP G+L D
Sbjct: 187 VDLLDPRVKSSLSEEEIIFPPYNAEELEDILTKRAQMAFKP---GVLPD----------- 232
Query: 661 WHSEKLAVCYGLISRRNESCIRIVKNLRICEDCHSFMK--LVSKVYQI---EIVVRDRTR 715
+ +C L +R + R + LR+ + MK V + Y E + RDR R
Sbjct: 233 ---NVIKLCAALAAREHGDARRALDLLRVSGEIAERMKDTKVKEEYVYMAKEEIERDRVR 289
>YPIA_BACSU (P54389) Hypothetical protein ypiA
Length = 423
Score = 32.3 bits (72), Expect = 4.6
Identities = 47/204 (23%), Positives = 87/204 (42%), Gaps = 14/204 (6%)
Query: 395 VNNALIDMYAKCGNLVKAREVFENMPRKN-----VISWSSMINAFAMHGNADSAIKLFRR 449
++ AL ++Y G KA + F+ + V + + + G + AI + +
Sbjct: 137 IDFALGELYFAQGAYAKAVQYFKTTAEEQSEIGGVNVHQRLAESLSASGEFEDAIPWYEK 196
Query: 450 MKEVNIEPNGVTFIGVLYACGHAGLVEEGEKLFSSMINEHGISPTREHYGCMVDLYCRAN 509
+ N +PN T G + AGLV+ K S + E S T +
Sbjct: 197 AVDENPDPN--TIFGYGFTALQAGLVKTAIKQLSDL-KELDPSYTSLYMPLSKSYEAEGM 253
Query: 510 F---LRKAIELIETMPFAPNVIIWGSLMSACQVHGEAELGEFAAKRLLELEPDHDGALVV 566
+ L+ A E I + + ++ + M A ++ G++E G+ + L L+P AL
Sbjct: 254 YEEALKTAKEGIRYDEYNKELFLYAAKM-ALKI-GKSEEGKKLLQEALALDPGFVEALHT 311
Query: 567 LSNIYAKEKRWND-VGLIRKSMSY 589
L +Y KE+ ++ + LI++ SY
Sbjct: 312 LLAVYHKEEDYDQIIDLIQEVRSY 335
>SEC8_SCHPO (O74562) Exocyst complex component sec8
Length = 1088
Score = 32.3 bits (72), Expect = 4.6
Identities = 43/195 (22%), Positives = 79/195 (40%), Gaps = 18/195 (9%)
Query: 498 YGCMVDLYCRANFLRKAIELIETMPFAPNVIIWGSLMSACQVHGEAELGEFAAKRLLELE 557
Y +++Y R + + I F + +G +M Q R++ L+
Sbjct: 90 YKSFLNVYDRISAALQTIAHTHKDDFTRGISAYGEIMEGIQK---------CNSRIIALK 140
Query: 558 PDHDGALVVLSNIYAKEKRWNDVGLIRKSMSYKGISKEKASSRI-EINNQVHMFMMADRY 616
+ + + N +KE + L R S K IS K + ++ + H + + +Y
Sbjct: 141 QSLEASQECIGNTNSKELQQT---LARSSQYKKVISVLKELNEANQLFDNFHTLVDSKQY 197
Query: 617 HKQSDEIYEKLDEVVSK-----LKLVGYKPSTSGILIDLEEEDKKELVLWHSEKLAVCYG 671
+ SD I DE+ L + +K +G+L LE+ +ELV K AV Y
Sbjct: 198 YHASDLIRRVWDELSRSDFDGILVVEQFKSRMTGLLSHLEDILSEELVSITFLKDAVAYP 257
Query: 672 LISRRNESCIRIVKN 686
++S + + +R N
Sbjct: 258 IVSYCSPNPLRETSN 272
>RA50_METKA (Q8TXI4) DNA double-strand break repair rad50 ATPase
Length = 876
Score = 32.3 bits (72), Expect = 4.6
Identities = 42/172 (24%), Positives = 76/172 (43%), Gaps = 18/172 (10%)
Query: 552 RLLELEPDH--------DGALVVLSNIYAKEKRWNDVGLIRKSMSYKGISKEKASSRIEI 603
R LE E DH +G L N+ K KR + RK + E A R+E
Sbjct: 585 RRLEEERDHVGQKLREAEGELERYHNLEEKVKRAREA---RKELKRIERDLEDAKGRLE- 640
Query: 604 NNQVHMFMMADRYHKQSDEIYEKLDEVVSKLKLVGYKPSTSGILIDLEEEDKKELVLWHS 663
+ ++ + +RY + D + E+L+ V K + V K S ++ E+ ++EL
Sbjct: 641 QVERNLEGLRERYGSE-DRLEEELESVEKKYERVRDKLSEVKGRLNGMEKRREEL----- 694
Query: 664 EKLAVCYGLISRRNESCIRIVKNLRICEDCHSFMKLVSKVYQIEIVVRDRTR 715
+K Y R E R+V+ L +C++ + + V++ + V R+ ++
Sbjct: 695 KKQVRKYREAKERKERLERVVEVLSLCKEVFRYSRDVAREKVLPAVEREASK 746
>CYCI_HUMAN (Q14094) Cyclin I
Length = 377
Score = 32.3 bits (72), Expect = 4.6
Identities = 29/108 (26%), Positives = 49/108 (44%), Gaps = 8/108 (7%)
Query: 17 ITTHPQLLTTLSSSTTLSHLKQIHAQILHSNTTPENTNTLLSKLALSICTLSSSS----- 71
++T PQLL +L + HL + Q+LH + S LAL++ +L
Sbjct: 159 VSTRPQLLFSLPKLSPSQHLAVLTKQLLHCMACNQLLQFRGSMLALAMVSLEMEKLIPDW 218
Query: 72 SSLHYALSVFSQIPNPH-THFSNQLLRHLS--RSSFPEKTIFLYHNLR 116
SL L +Q+ + H + HLS +SS P ++++Y L+
Sbjct: 219 LSLTIELLQKAQMDSSQLIHCRELVAHHLSTLQSSLPLNSVYVYRPLK 266
>DUT_HHV6U (Q06095) Deoxyuridine 5'-triphosphate nucleotidohydrolase
(EC 3.6.1.23) (dUTPase) (dUTP pyrophosphatase)
Length = 376
Score = 32.0 bits (71), Expect = 6.0
Identities = 18/91 (19%), Positives = 35/91 (37%), Gaps = 4/91 (4%)
Query: 381 HTYVDRSGFGRALSVNNALIDMYAKCGNLVKAREVFENMPRKNVISWSSMINAFAMHGNA 440
H R + S D C + +KA + P K + +I + + +
Sbjct: 257 HLKASREFIAKPNSYTIQYFDAMYVCADELKALMI----PSKEIAKLGLLIETYIWNKDT 312
Query: 441 DSAIKLFRRMKEVNIEPNGVTFIGVLYACGH 471
+IK+F ++ P G+ +++ CGH
Sbjct: 313 IPSIKIFNSTRKTIYIPTGICIARIIFTCGH 343
>DUT_HHV6G (P30007) Deoxyuridine 5'-triphosphate nucleotidohydrolase
(EC 3.6.1.23) (dUTPase) (dUTP pyrophosphatase)
Length = 376
Score = 32.0 bits (71), Expect = 6.0
Identities = 18/91 (19%), Positives = 35/91 (37%), Gaps = 4/91 (4%)
Query: 381 HTYVDRSGFGRALSVNNALIDMYAKCGNLVKAREVFENMPRKNVISWSSMINAFAMHGNA 440
H R + S D C + +KA + P K + +I + + +
Sbjct: 257 HLKASREFIAKPNSYTIQYFDAMYVCADELKALMI----PSKEIAKLGLLIETYIWNKDT 312
Query: 441 DSAIKLFRRMKEVNIEPNGVTFIGVLYACGH 471
+IK+F ++ P G+ +++ CGH
Sbjct: 313 IPSIKIFNSTRKTIYIPTGICIARIIFTCGH 343
>DPO1_RICHE (Q9RLB6) DNA polymerase I (EC 2.7.7.7) (POL I)
Length = 921
Score = 32.0 bits (71), Expect = 6.0
Identities = 29/104 (27%), Positives = 42/104 (39%), Gaps = 18/104 (17%)
Query: 389 FGRALSVNNALIDMYAKCGNLVKAREVFENMPRKNVISWSSMINAFAMHGNADSAIKLFR 448
FG ++ N+L + + VK RE +N +ISW + + N D +L
Sbjct: 206 FGSVENIFNSLDQVSS-----VKQRETLQNSREAALISWQLI----GLDSNVDLDFQLN- 255
Query: 449 RMKEVNIE---PNGVTFIGVLYACGHAGLVEEGEKLFSSMINEH 489
N+E PN G L G L + E LF IN+H
Sbjct: 256 -----NLEWSPPNSDKLTGFLQEYGFRSLYKRAENLFDIKINDH 294
>PSD3_DAUCA (Q06364) Probable 26S proteasome non-ATPase regulatory
subunit 3 (26S proteasome subunit S3) (Nuclear antigen
21D7)
Length = 489
Score = 31.6 bits (70), Expect = 7.8
Identities = 19/70 (27%), Positives = 32/70 (45%), Gaps = 1/70 (1%)
Query: 21 PQLLTTLSSSTTLSHLKQIHAQILHSNTTPENTNTLLSKLALSICTLSSSSSSLHYALSV 80
P T ++ +TL HLK+I A ++ S +L + L+I ++S+ A
Sbjct: 15 PSNSVTAATPSTLQHLKEI-ASLIESGAYAREVRRILRAVRLTIALRKKLNASVVNAFLN 73
Query: 81 FSQIPNPHTH 90
FS +P H
Sbjct: 74 FSLVPGSEVH 83
>NASD_BACSU (P42435) Nitrite reductase [NAD(P)H] (EC 1.7.1.4)
Length = 805
Score = 31.6 bits (70), Expect = 7.8
Identities = 18/65 (27%), Positives = 32/65 (48%)
Query: 498 YGCMVDLYCRANFLRKAIELIETMPFAPNVIIWGSLMSACQVHGEAELGEFAAKRLLELE 557
YG + LY +A L K + IET P+ +V+ +S +V + E K+ +++
Sbjct: 286 YGLVAPLYEQAKVLAKHMCGIETKPYEGSVLSTQLKVSGVEVFSAGDFNESEEKKAIKVF 345
Query: 558 PDHDG 562
+ DG
Sbjct: 346 DEQDG 350
Database: sprot
Posted date: Nov 25, 2004 10:54 AM
Number of letters in database: 59,974,054
Number of sequences in database: 164,201
Lambda K H
0.322 0.135 0.403
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 81,514,153
Number of Sequences: 164201
Number of extensions: 3320159
Number of successful extensions: 8912
Number of sequences better than 10.0: 20
Number of HSP's better than 10.0 without gapping: 4
Number of HSP's successfully gapped in prelim test: 16
Number of HSP's that attempted gapping in prelim test: 8886
Number of HSP's gapped (non-prelim): 39
length of query: 730
length of database: 59,974,054
effective HSP length: 118
effective length of query: 612
effective length of database: 40,598,336
effective search space: 24846181632
effective search space used: 24846181632
T: 11
A: 40
X1: 16 ( 7.4 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 41 (22.0 bits)
S2: 70 (31.6 bits)
Medicago: description of AC135320.10