
BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query= BAB33201.1 510 aa
(510 letters)
Database: ara_mips
26,719 sequences; 11,318,596 total letters
Searching..................................................done
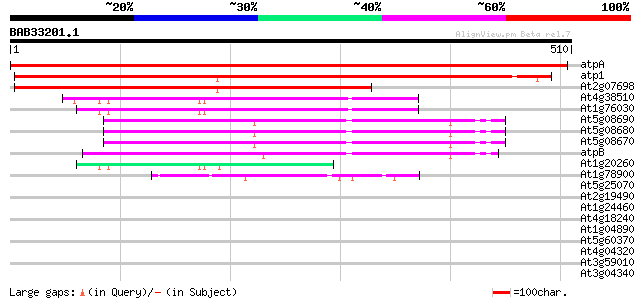
Score E
Sequences producing significant alignments: (bits) Value
atpA -chloroplast genome- ATPase alpha subunit 907 0.0
atp1 -mitochondrial genome- ATPase subunit 1 551 e-157
At2g07698 putative protein 397 e-110
At4g38510 vacuolar-type H+-ATPase subunit B2 (VHA-B2) 124 2e-28
At1g76030 vacuolar-type H+-ATPase subunit B1 (VHA-B1) 122 4e-28
At5g08690 H+-transporting ATP synthase beta chain (mitochondrial... 96 4e-20
At5g08680 H+-transporting ATP synthase beta chain (mitochondrial... 96 4e-20
At5g08670 H+-transporting ATP synthase beta chain (mitochondrial... 96 4e-20
atpB -chloroplast genome- ATPase beta subunit 94 1e-19
At1g20260 vacuolar-type H+-ATPase subunit B3 (VHA-B3) 85 1e-16
At1g78900 vacuolar-type H+-ATPase subunit A (VHA-A) 80 3e-15
At5g25070 unknown protein 37 0.034
At2g19490 putative recA protein 31 1.9
At1g24460 unknown protein 31 1.9
At4g18240 starch synthase-like protein 30 2.4
At1g04890 hypothetical protein 30 3.2
At5g60370 unknown protein (At5g60370) 29 5.4
At4g04320 malonyl-CoA decarboxylase like protein 29 7.0
At3g59010 pectinesterase precursor-like protein 29 7.0
At3g04340 unknown protein 29 7.0
>atpA -chloroplast genome- ATPase alpha subunit
Length = 507
Score = 907 bits (2345), Expect = 0.0
Identities = 474/507 (93%), Positives = 491/507 (96%)
Query: 1 MVTIRADEISKIIRERIEQYNTEIKIVNTGTVLQVGDGIARIYGLDEVMAGELVEFEEGT 60
MVTIRADEIS IIRERIEQYN E+ IVNTGTVLQVGDGIARIYGLDEVMAGELVEFEEGT
Sbjct: 1 MVTIRADEISNIIRERIEQYNREVTIVNTGTVLQVGDGIARIYGLDEVMAGELVEFEEGT 60
Query: 61 IGIALNLESKNVGVVLMGDGLMIQEGSSVKATGRIAQIPVSEGYLGRVINALAKPIDGRG 120
IGIALNLES NVGVVLMGDGLMIQEGSSVKATG+IAQIPVSE YLGRVINALA PIDGRG
Sbjct: 61 IGIALNLESNNVGVVLMGDGLMIQEGSSVKATGKIAQIPVSEAYLGRVINALANPIDGRG 120
Query: 121 EISSSESRLIESPAPGIISRRSVYEPLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVA 180
+IS+SESRLIESPAPGIISRRSVYEPLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVA
Sbjct: 121 KISASESRLIESPAPGIISRRSVYEPLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVA 180
Query: 181 TDTILNQQGQNVICVYVAVGQKASSVAQVVNTLQERGAMEYTIVVAETADSPATLQYLAP 240
TDTILNQQGQNVICVYVA+GQKASSVAQVV +LQERGAMEYTIVVAETADSPATLQYLAP
Sbjct: 181 TDTILNQQGQNVICVYVAIGQKASSVAQVVTSLQERGAMEYTIVVAETADSPATLQYLAP 240
Query: 241 YTGAALAEFFMYRERHTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLHSRLLE 300
YTGAALAE+FMYRE+HTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLHSRLLE
Sbjct: 241 YTGAALAEYFMYREQHTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLHSRLLE 300
Query: 301 RAAKLSSQLGEGSMTALPIVETQSGDVSAYIPTNVISITDGQIFLSADLFNAGIRPAINV 360
RAAKLSSQLGEGSMTALPIVETQSGDVSAYIPTNVISITDGQIFLSADLFNAGIRPAINV
Sbjct: 301 RAAKLSSQLGEGSMTALPIVETQSGDVSAYIPTNVISITDGQIFLSADLFNAGIRPAINV 360
Query: 361 GISVSRVGSAAQIKAMKQVAGKLKLELAQFAELEAFAQFASDLDKATQNQLARGQRLREL 420
GISVSRVGSAAQIKAMKQVAGKLKLELAQFAELEAF+QF+SDLDKATQNQLARGQRLREL
Sbjct: 361 GISVSRVGSAAQIKAMKQVAGKLKLELAQFAELEAFSQFSSDLDKATQNQLARGQRLREL 420
Query: 421 LKQSQSAPLTVEEQVITIYTGTNGYLDSLEIRQVRKFLVELRAYLKTNKPQFNEIISSTK 480
LKQSQSAPLTVEEQ++TIYTGTNGYLD LEI QVRKFLV+LR YLKTNKPQF EII+STK
Sbjct: 421 LKQSQSAPLTVEEQIMTIYTGTNGYLDGLEIGQVRKFLVQLRTYLKTNKPQFQEIIASTK 480
Query: 481 TFTGEAEALLKEAIQEQMELFLLQEQV 507
T T EAE+ LKE IQEQ+E FLLQE+V
Sbjct: 481 TLTAEAESFLKEGIQEQLERFLLQEKV 507
>atp1 -mitochondrial genome- ATPase subunit 1
Length = 507
Score = 551 bits (1419), Expect = e-157
Identities = 284/499 (56%), Positives = 362/499 (71%), Gaps = 14/499 (2%)
Query: 5 RADEISKIIRERIEQYNTEIKIVNTGTVLQVGDGIARIYGLDEVMAGELVEFEEGTIGIA 64
RA E++ + RI + ++ G V+ VGDGIA++YGL+E+ AGE+V F G G+A
Sbjct: 6 RAAELTNLFESRIRNFYANFQVDEIGRVVSVGDGIAQVYGLNEIQAGEMVLFANGVKGMA 65
Query: 65 LNLESKNVGVVLMGDGLMIQEGSSVKATGRIAQIPVSEGYLGRVINALAKPIDGRGEISS 124
LNLE++NVG+V+ G I+EG VK TG I +P + LGRV++A+ PIDG+G +S
Sbjct: 66 LNLENENVGIVVFGGDTAIKEGDLVKRTGSIVDVPAGKAMLGRVVDAMGVPIDGKGALSD 125
Query: 125 SESRLIESPAPGIISRRSVYEPLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVATDTI 184
E R +E APGI+ R+SV+EP+QTGL A+DS++PIGRGQREL+IG RQTGKT +A DTI
Sbjct: 126 HEQRRVEVKAPGILERKSVHEPMQTGLKAVDSLVPIGRGQRELLIGGRQTGKTTIAIDTI 185
Query: 185 LNQ---------QGQNVICVYVAVGQKASSVAQVVNTLQERGAMEYTIVVAETADSPATL 235
LNQ + + + CVYVA+GQK S+V Q++ TL+E A+EY+I+VA TA PA L
Sbjct: 186 LNQKQINSRATSESETMYCVYVAIGQKRSTVGQLIQTLEEANALEYSILVAATASDPAPL 245
Query: 236 QYLAPYTGAALAEFFMYRERHTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLH 295
Q+LAPY+G A+ E+F H LIIYDDLSKQA AYRQMSLLLRRPPGREA+PGDVFYLH
Sbjct: 246 QFLAPYSGCAMGEYFRDNGMHALIIYDDLSKQAVAYRQMSLLLRRPPGREAFPGDVFYLH 305
Query: 296 SRLLERAAKLSSQLGEGSMTALPIVETQSGDVSAYIPTNVISITDGQIFLSADLFNAGIR 355
SRLLERAAK S Q G GS+TALP++ETQ+GDVSAYIPTNVISITDGQI L +LF GIR
Sbjct: 306 SRLLERAAKRSDQTGAGSLTALPVIETQAGDVSAYIPTNVISITDGQICLETELFYRGIR 365
Query: 356 PAINVGISVSRVGSAAQIKAMKQVAGKLKLELAQFAELEAFAQFASDLDKATQNQLARGQ 415
PAINVG+SVSRVGSAAQ+KAMKQV G KLELAQ+ E+ AFAQF SDLD ATQ L RG
Sbjct: 366 PAINVGLSVSRVGSAAQLKAMKQVCGSSKLELAQYREVAAFAQFGSDLDAATQALLNRGA 425
Query: 416 RLRELLKQSQSAPLTVEEQVITIYTGTNGYLDSLEIRQVRKFLVELRAYLKTNKPQFNEI 475
RL E+ KQ Q APL +E+Q++ IY NG+ D + + ++ ++ +A + KP+ +
Sbjct: 426 RLTEVPKQPQYAPLPIEKQILVIYAAVNGFCDRMPLDRISQY---EKAIPNSVKPELLQA 482
Query: 476 ISS--TKTFTGEAEALLKE 492
+ T E +A LKE
Sbjct: 483 LKGGLTNERKMEPDAFLKE 501
>At2g07698 putative protein
Length = 609
Score = 397 bits (1019), Expect = e-110
Identities = 195/334 (58%), Positives = 250/334 (74%), Gaps = 9/334 (2%)
Query: 5 RADEISKIIRERIEQYNTEIKIVNTGTVLQVGDGIARIYGLDEVMAGELVEFEEGTIGIA 64
RA E++ + RI + ++ G V+ VGDGIA++YGL+E+ AGE+V F G G+A
Sbjct: 276 RAAELTNLFESRIRNFYANFQVDEIGRVVSVGDGIAQVYGLNEIQAGEMVLFANGVKGMA 335
Query: 65 LNLESKNVGVVLMGDGLMIQEGSSVKATGRIAQIPVSEGYLGRVINALAKPIDGRGEISS 124
LNLE++NVG+V+ G I+EG VK TG I +P + LGRV++A+ PIDG+G +S
Sbjct: 336 LNLENENVGIVVFGGDTAIKEGDLVKRTGSIVDVPAGKAMLGRVVDAMGVPIDGKGALSD 395
Query: 125 SESRLIESPAPGIISRRSVYEPLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVATDTI 184
E R +E APGI+ R+SV+EP+QTGL A+DS++PIGRGQREL+IGDRQTGKT +A DTI
Sbjct: 396 HEQRRVEVKAPGILERKSVHEPMQTGLKAVDSLVPIGRGQRELLIGDRQTGKTTIAIDTI 455
Query: 185 LNQ---------QGQNVICVYVAVGQKASSVAQVVNTLQERGAMEYTIVVAETADSPATL 235
LNQ + + + CVYVA+GQK S+V Q++ TL+E A+EY+I+VA TA PA L
Sbjct: 456 LNQKQINSRATSESETMYCVYVAIGQKRSTVGQLIQTLEEANALEYSILVAATASDPAPL 515
Query: 236 QYLAPYTGAALAEFFMYRERHTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLH 295
Q+LAPY+G A+ E+F H LIIYDDLSKQA AYRQMSLLLRRPPGREA+PGDVFYLH
Sbjct: 516 QFLAPYSGCAMGEYFRDNGMHALIIYDDLSKQAVAYRQMSLLLRRPPGREAFPGDVFYLH 575
Query: 296 SRLLERAAKLSSQLGEGSMTALPIVETQSGDVSA 329
SRLLERAAK S Q G GS+TALP++ETQ+GDVSA
Sbjct: 576 SRLLERAAKRSDQTGAGSLTALPVIETQAGDVSA 609
>At4g38510 vacuolar-type H+-ATPase subunit B2 (VHA-B2)
Length = 487
Score = 124 bits (310), Expect = 2e-28
Identities = 105/359 (29%), Positives = 160/359 (44%), Gaps = 38/359 (10%)
Query: 49 MAGELVEFE-------EGTIGIALNLESKNVGVVLMGDG----LMIQEGSS--------V 89
+AG LV E + + I L + G VL DG + + EG+S V
Sbjct: 26 VAGPLVILEKVKGPKYQEIVNIRLGDGTTRRGQVLEVDGEKAVVQVFEGTSGIDNKYTTV 85
Query: 90 KATGRIAQIPVSEGYLGRVINALAKPIDGRGEISSSESRLIESPAPGIISRRSVYEPLQT 149
+ TG + + PVS LGR+ N KPID I I + R E +QT
Sbjct: 86 QFTGEVLKTPVSLDMLGRIFNGSGKPIDNGPPILPEAYLDISGSSINPSERTYPEEMIQT 145
Query: 150 GLIAIDSMIPIGRGQRELIIGD-------------RQTG--KTAVATDTIL-NQQGQNVI 193
G+ ID M I RGQ+ + RQ G K +D +L +Q+ N
Sbjct: 146 GISTIDVMNSIARGQKIPLFSAAGLPHNEIAAQICRQAGLVKRLEKSDNLLEHQEDDNFA 205
Query: 194 CVYVAVGQKASSVAQVVNTLQERGAMEYTIVVAETADSPATLQYLAPYTGAALAEFFMYR 253
V+ A+G + +E G+ME + A+ P + + P AE+ Y
Sbjct: 206 IVFAAMGVNMETAQFFKRDFEENGSMERVTLFLNLANDPTIERIITPRIALTTAEYLAYE 265
Query: 254 -ERHTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLHSRLLERAAKLSSQLGEG 312
+H L+I D+S A A R++S PGR YPG ++ + + ERA ++ + +G
Sbjct: 266 CGKHVLVILTDMSSYADALREVSAAREEVPGRRGYPGYMYTDLATIYERAGRIEGR--KG 323
Query: 313 SMTALPIVETQSGDVSAYIPTNVISITDGQIFLSADLFNAGIRPAINVGISVSRVGSAA 371
S+T +PI+ + D++ P IT+GQI++ L N I P INV S+SR+ +A
Sbjct: 324 SITQIPILTMPNDDITHPTPDLTGYITEGQIYIDRQLHNRQIYPPINVLPSLSRLMKSA 382
>At1g76030 vacuolar-type H+-ATPase subunit B1 (VHA-B1)
Length = 486
Score = 122 bits (307), Expect = 4e-28
Identities = 101/340 (29%), Positives = 151/340 (43%), Gaps = 31/340 (9%)
Query: 61 IGIALNLESKNVGVVLMGDG----LMIQEGSS--------VKATGRIAQIPVSEGYLGRV 108
+ I L S G VL DG + + EG+S V+ TG + + PVS LGR+
Sbjct: 44 VNIRLGDGSTRRGQVLEVDGEKAVVQVFEGTSGIDNKFTTVQFTGEVLKTPVSLDMLGRI 103
Query: 109 INALAKPIDGRGEISSSESRLIESPAPGIISRRSVYEPLQTGLIAIDSMIPIGRGQRELI 168
N KPID I I + R E +QTG+ ID M I RGQ+ +
Sbjct: 104 FNGSGKPIDNGPPILPEAYLDISGSSINPSERTYPEEMIQTGISTIDVMNSIARGQKIPL 163
Query: 169 IGD-------------RQTG--KTAVATDTILNQQGQ-NVICVYVAVGQKASSVAQVVNT 212
RQ G K T +L G+ N V+ A+G +
Sbjct: 164 FSAAGLPHNEIAAQICRQAGLVKRLEKTVDLLEDHGEDNFAIVFAAMGVNMETAQFFKRD 223
Query: 213 LQERGAMEYTIVVAETADSPATLQYLAPYTGAALAEFFMYR-ERHTLIIYDDLSKQAQAY 271
+E G+ME + A+ P + + P AE+ Y +H L+I D+S A A
Sbjct: 224 FEENGSMERVTLFLNLANDPTIERIITPRIALTTAEYLAYECGKHVLVILTDMSSYADAL 283
Query: 272 RQMSLLLRRPPGREAYPGDVFYLHSRLLERAAKLSSQLGEGSMTALPIVETQSGDVSAYI 331
R++S PGR YPG ++ + + ERA ++ + +GS+T +PI+ + D++
Sbjct: 284 REVSAAREEVPGRRGYPGYMYTDLATIYERAGRIEGR--KGSITQIPILTMPNDDITHPT 341
Query: 332 PTNVISITDGQIFLSADLFNAGIRPAINVGISVSRVGSAA 371
P IT+GQI++ L N I P INV S+SR+ +A
Sbjct: 342 PDLTGYITEGQIYIDRQLHNRQIYPPINVLPSLSRLMKSA 381
>At5g08690 H+-transporting ATP synthase beta chain (mitochondrial)
-like protein
Length = 556
Score = 96.3 bits (238), Expect = 4e-20
Identities = 95/379 (25%), Positives = 160/379 (42%), Gaps = 26/379 (6%)
Query: 86 GSSVKATGRIAQIPVSEGYLGRVINALAKPIDGRGEISSSESRLIESPAPGIISRRSVYE 145
G V TG +PV LGR++N L +PID RGEI + I AP ++ + E
Sbjct: 147 GRKVLNTGAPITVPVGRATLGRIMNVLGEPIDERGEIKTEHYLPIHRDAPALVDLATGQE 206
Query: 146 PLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVATDTILN-QQGQNVICVYVAVGQKAS 204
L TG+ +D + P RG + + G GKT + + I N + V+ VG++
Sbjct: 207 ILATGIKVVDLLAPYQRGGKIGLFGGAGVGKTVLIMELINNVAKAHGGFSVFAGVGERTR 266
Query: 205 SVAQVVNTLQERGAMEY--------TIVVAETADSPATLQYLAPYTGAALAEFFMYRE-R 255
+ + E G ++ +V + P + TG +AE+F E +
Sbjct: 267 EGNDLYREMIESGVIKLGEKQSESKCALVYGQMNEPPGARARVGLTGLTVAEYFRDAEGQ 326
Query: 256 HTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLHSRLLERAAKLSSQLGEGSMT 315
L+ D++ + QA ++S LL R P Y + L ER +GS+T
Sbjct: 327 DVLLFIDNIFRFTQANSEVSALLGRIPSAVGYQPTLASDLGALQERITTTK----KGSIT 382
Query: 316 ALPIVETQSGDVSAYIPTNVISITDGQIFLSADLFNAGIRPAINVGISVSRVGSAAQI-K 374
++ + + D++ P + D LS + GI PA++ S SR+ S + +
Sbjct: 383 SVQAIYVPADDLTDPAPATTFAHLDATTVLSRQISELGIYPAVDPLDSTSRMLSPHILGE 442
Query: 375 AMKQVAGKLKLELAQFAELEAFAQF--ASDLDKATQNQLARGQRLRELLKQSQSAPLTVE 432
A ++ L + L+ +L + + +AR ++++ L Q P V
Sbjct: 443 EHYNTARGVQKVLQNYKNLQDIIAILGMDELSEDDKLTVARARKIQRFLSQ----PFHVA 498
Query: 433 EQVITIYTGTNG-YLDSLE 450
E I+TG G Y+D E
Sbjct: 499 E----IFTGAPGKYVDLKE 513
>At5g08680 H+-transporting ATP synthase beta chain (mitochondrial)
-like protein
Length = 559
Score = 96.3 bits (238), Expect = 4e-20
Identities = 95/379 (25%), Positives = 160/379 (42%), Gaps = 26/379 (6%)
Query: 86 GSSVKATGRIAQIPVSEGYLGRVINALAKPIDGRGEISSSESRLIESPAPGIISRRSVYE 145
G V TG +PV LGR++N L +PID RGEI + I AP ++ + E
Sbjct: 150 GRKVLNTGAPITVPVGRATLGRIMNVLGEPIDERGEIKTEHYLPIHRDAPALVDLATGQE 209
Query: 146 PLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVATDTILN-QQGQNVICVYVAVGQKAS 204
L TG+ +D + P RG + + G GKT + + I N + V+ VG++
Sbjct: 210 ILATGIKVVDLLAPYQRGGKIGLFGGAGVGKTVLIMELINNVAKAHGGFSVFAGVGERTR 269
Query: 205 SVAQVVNTLQERGAMEY--------TIVVAETADSPATLQYLAPYTGAALAEFFMYRE-R 255
+ + E G ++ +V + P + TG +AE+F E +
Sbjct: 270 EGNDLYREMIESGVIKLGEKQSESKCALVYGQMNEPPGARARVGLTGLTVAEYFRDAEGQ 329
Query: 256 HTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLHSRLLERAAKLSSQLGEGSMT 315
L+ D++ + QA ++S LL R P Y + L ER +GS+T
Sbjct: 330 DVLLFIDNIFRFTQANSEVSALLGRIPSAVGYQPTLASDLGALQERITTTK----KGSIT 385
Query: 316 ALPIVETQSGDVSAYIPTNVISITDGQIFLSADLFNAGIRPAINVGISVSRVGSAAQI-K 374
++ + + D++ P + D LS + GI PA++ S SR+ S + +
Sbjct: 386 SVQAIYVPADDLTDPAPATTFAHLDATTVLSRQISELGIYPAVDPLDSTSRMLSPHILGE 445
Query: 375 AMKQVAGKLKLELAQFAELEAFAQF--ASDLDKATQNQLARGQRLRELLKQSQSAPLTVE 432
A ++ L + L+ +L + + +AR ++++ L Q P V
Sbjct: 446 EHYNTARGVQKVLQNYKNLQDIIAILGMDELSEDDKLTVARARKIQRFLSQ----PFHVA 501
Query: 433 EQVITIYTGTNG-YLDSLE 450
E I+TG G Y+D E
Sbjct: 502 E----IFTGAPGKYVDLKE 516
>At5g08670 H+-transporting ATP synthase beta chain (mitochondrial)
-like protein
Length = 556
Score = 96.3 bits (238), Expect = 4e-20
Identities = 95/379 (25%), Positives = 160/379 (42%), Gaps = 26/379 (6%)
Query: 86 GSSVKATGRIAQIPVSEGYLGRVINALAKPIDGRGEISSSESRLIESPAPGIISRRSVYE 145
G V TG +PV LGR++N L +PID RGEI + I AP ++ + E
Sbjct: 147 GRKVLNTGAPITVPVGRATLGRIMNVLGEPIDERGEIKTEHYLPIHRDAPALVDLATGQE 206
Query: 146 PLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVATDTILN-QQGQNVICVYVAVGQKAS 204
L TG+ +D + P RG + + G GKT + + I N + V+ VG++
Sbjct: 207 ILATGIKVVDLLAPYQRGGKIGLFGGAGVGKTVLIMELINNVAKAHGGFSVFAGVGERTR 266
Query: 205 SVAQVVNTLQERGAMEY--------TIVVAETADSPATLQYLAPYTGAALAEFFMYRE-R 255
+ + E G ++ +V + P + TG +AE+F E +
Sbjct: 267 EGNDLYREMIESGVIKLGEKQSESKCALVYGQMNEPPGARARVGLTGLTVAEYFRDAEGQ 326
Query: 256 HTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLHSRLLERAAKLSSQLGEGSMT 315
L+ D++ + QA ++S LL R P Y + L ER +GS+T
Sbjct: 327 DVLLFIDNIFRFTQANSEVSALLGRIPSAVGYQPTLASDLGALQERITTTK----KGSIT 382
Query: 316 ALPIVETQSGDVSAYIPTNVISITDGQIFLSADLFNAGIRPAINVGISVSRVGSAAQI-K 374
++ + + D++ P + D LS + GI PA++ S SR+ S + +
Sbjct: 383 SVQAIYVPADDLTDPAPATTFAHLDATTVLSRQISELGIYPAVDPLDSTSRMLSPHILGE 442
Query: 375 AMKQVAGKLKLELAQFAELEAFAQF--ASDLDKATQNQLARGQRLRELLKQSQSAPLTVE 432
A ++ L + L+ +L + + +AR ++++ L Q P V
Sbjct: 443 EHYNTARGVQKVLQNYKNLQDIIAILGMDELSEDDKLTVARARKIQRFLSQ----PFHVA 498
Query: 433 EQVITIYTGTNG-YLDSLE 450
E I+TG G Y+D E
Sbjct: 499 E----IFTGAPGKYVDLKE 513
>atpB -chloroplast genome- ATPase beta subunit
Length = 498
Score = 94.4 bits (233), Expect = 1e-19
Identities = 91/390 (23%), Positives = 163/390 (41%), Gaps = 24/390 (6%)
Query: 67 LESKNVGVVLMGDGLMIQEGSSVKATGRIAQIPVSEGYLGRVINALAKPIDGRGEISSSE 126
L + V V M ++ G V G +PV LGR+ N L +P+D G + +
Sbjct: 69 LGNNRVRAVAMSATEGLKRGMDVVDMGNPLSVPVGGATLGRIFNVLGEPVDNLGPVDTRT 128
Query: 127 SRLIESPAPGIISRRSVYEPLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVATDTILN 186
+ I AP I + +TG+ +D + P RG + + G GKT + + I N
Sbjct: 129 TSPIHKSAPAFIELDTKLSIFETGIKVVDLLAPYRRGGKIGLFGGAGVGKTVLIMELINN 188
Query: 187 -QQGQNVICVYVAVGQKASSVAQVVNTLQERGAM-EYTIVVAETA------DSPATLQYL 238
+ + V+ VG++ + ++E G + E + ++ A + P +
Sbjct: 189 IAKAHGGVSVFGGVGERTREGNDLYMEMKESGVINEQNLAESKVALVYGQMNEPPGARMR 248
Query: 239 APYTGAALAEFFM-YRERHTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLHSR 297
T +AE+F E+ L+ D++ + QA ++S LL R P Y +
Sbjct: 249 VGLTALTMAEYFRDVNEQDVLLFIDNIFRFVQAGSEVSALLGRMPSAVGYQPTLSTEMGT 308
Query: 298 LLERAAKLSSQLGEGSMTALPIVETQSGDVSAYIPTNVISITDGQIFLSADLFNAGIRPA 357
L ER +GS+T++ V + D++ P + D LS L GI PA
Sbjct: 309 LQERITSTK----KGSITSIQAVYVPADDLTDPAPATTFAHLDATTVLSRGLAAKGIYPA 364
Query: 358 INVGISVSRVGSAAQI-KAMKQVAGKLKLELAQFAELEAFAQF--ASDLDKATQNQLARG 414
++ S S + + + + A ++K L ++ EL+ +L + + +AR
Sbjct: 365 VDPLDSTSTMLQPRIVGEEHYETAQQVKQTLQRYKELQDIIAILGLDELSEEDRLTVARA 424
Query: 415 QRLRELLKQSQSAPLTVEEQVITIYTGTNG 444
+++ L Q P V E ++TG+ G
Sbjct: 425 RKIERFLSQ----PFFVAE----VFTGSPG 446
>At1g20260 vacuolar-type H+-ATPase subunit B3 (VHA-B3)
Length = 330
Score = 84.7 bits (208), Expect = 1e-16
Identities = 73/264 (27%), Positives = 107/264 (39%), Gaps = 30/264 (11%)
Query: 61 IGIALNLESKNVGVVLMGDG----LMIQEGSS--------VKATGRIAQIPVSEGYLGRV 108
+ I L S G VL DG + + EG+S V+ TG + + PVS LGR+
Sbjct: 44 VNIRLGDGSTRRGQVLEVDGEKAVVQVFEGTSGIDNKFTTVQFTGEVLKTPVSLDMLGRI 103
Query: 109 INALAKPIDGRGEISSSESRLIESPAPGIISRRSVYEPLQTGLIAIDSMIPIGRGQRELI 168
N KPID I I + R E +QTG+ ID M I RGQ+ +
Sbjct: 104 FNGSGKPIDNGPPILPEAYLDISGSSINPSERTYPEEMIQTGISTIDVMNSIARGQKIPL 163
Query: 169 IGD-------------RQTG--KTAVATDTILNQQG--QNVICVYVAVGQKASSVAQVVN 211
RQ G K T+ ++ + N V+ A+G +
Sbjct: 164 FSAAGLPHNEIAAQICRQAGLVKRLEKTENLIQEDHGEDNFAIVFAAMGVNMETAQFFKR 223
Query: 212 TLQERGAMEYTIVVAETADSPATLQYLAPYTGAALAEFFMYR-ERHTLIIYDDLSKQAQA 270
+E G+ME + A+ P + + P AE+ Y +H L+I D+S A A
Sbjct: 224 DFEENGSMERVTLFLNLANDPTIERIITPRIALTTAEYLAYECGKHVLVILTDMSSYADA 283
Query: 271 YRQMSLLLRRPPGREAYPGDVFYL 294
R++S PGR YPG + +
Sbjct: 284 LREVSAAREEVPGRRGYPGYIIQI 307
>At1g78900 vacuolar-type H+-ATPase subunit A (VHA-A)
Length = 623
Score = 79.7 bits (195), Expect = 3e-15
Identities = 73/261 (27%), Positives = 121/261 (45%), Gaps = 26/261 (9%)
Query: 130 IESPAPGIISRRSVYEPLQTGLIAIDSMIPIGRGQRELIIGDRQTGKTAVATDTILNQQG 189
+ +P P + S+ + PL TG +D++ P G I G GKT ++ L++
Sbjct: 213 VRTPRP-VASKLAADTPLLTGQRVLDALFPSVLGGTCAIPGAFGCGKTVISQ--ALSKYS 269
Query: 190 QNVICVYVAVGQKASSVAQVVNTL----------QERGAMEYTIVVAETADSPATLQYLA 239
+ VYV G++ + +A+V+ +E M+ T +VA T++ P + +
Sbjct: 270 NSDAVVYVGCGERGNEMAEVLMDFPQLTMTLPDGREESVMKRTTLVANTSNMPVAAREAS 329
Query: 240 PYTGAALAEFFMYRERHTLIIYDDLSKQAQAYRQMSLLLRRPPGREAYPGDVFYLHSRL- 298
YTG +AE+F + ++ D S+ A+A R++S L P YP YL +RL
Sbjct: 330 IYTGITIAEYFRDMGYNVSMMADSTSRWAEALREISGRLAEMPADSGYPA---YLAARLA 386
Query: 299 --LERAAKLSSQLG---EGSMTALPIVETQSGDVSAYIPTNVISITDGQIFLSAD--LFN 351
ERA K+ G GS+T + V GD S + + +SI Q+F D L
Sbjct: 387 SFYERAGKVKCLGGPERNGSVTIVGAVSPPGGDFSDPVTSATLSIV--QVFWGLDKKLAQ 444
Query: 352 AGIRPAINVGISVSRVGSAAQ 372
P++N IS S+ +A +
Sbjct: 445 RKHFPSVNWLISYSKYSTALE 465
>At5g25070 unknown protein
Length = 736
Score = 36.6 bits (83), Expect = 0.034
Identities = 30/111 (27%), Positives = 51/111 (45%), Gaps = 4/111 (3%)
Query: 392 ELEAFAQFASDLDKATQNQLARGQRLRELLKQSQSAPLTVEEQVITIYTGTNGYLDSLEI 451
E E ++ D+D+ ++ RG +LR+L + S EE VI + G Y+ +
Sbjct: 452 ETEDLSRKKKDVDEFMTSEKERGAKLRDLARVSADEACEYEE-VIKLRKGLMSYVS--KT 508
Query: 452 RQVRKFLVELRAYLKTNKPQFNEIISSTKTFTGEAEALLKEAIQEQMELFL 502
R+ R LV + L + E +SST+ E + K IQ+ + F+
Sbjct: 509 REERAKLVNIEEKLSEEVQKLQEEVSSTRELLKERSS-KKSIIQQNITSFM 558
>At2g19490 putative recA protein
Length = 376
Score = 30.8 bits (68), Expect = 1.9
Identities = 23/71 (32%), Positives = 34/71 (47%), Gaps = 4/71 (5%)
Query: 149 TGLIAIDSMIPIG---RGQRELIIGDRQTGKTAVATDTILNQQGQNVICVYV-AVGQKAS 204
TG A+D + +G +G+ I G +GKT +A I Q Q CV+V A S
Sbjct: 41 TGSFALDVALGVGGLPKGRVVEIYGPEASGKTTLALHVIAEAQKQGGTCVFVDAEHALDS 100
Query: 205 SVAQVVNTLQE 215
S+A+ + E
Sbjct: 101 SLAKAIGVNTE 111
>At1g24460 unknown protein
Length = 1791
Score = 30.8 bits (68), Expect = 1.9
Identities = 37/146 (25%), Positives = 67/146 (45%), Gaps = 10/146 (6%)
Query: 364 VSRVGSAAQIKAMKQVAGKLKLELAQFAELEAFAQFASDLDKATQNQLARGQRLRELLKQ 423
+S G+AA+ + +V L LEL QF E E + S D + QR++EL
Sbjct: 1283 ISACGAAAR-ELQLEVKNNL-LELVQFQENENGGEMESTEDPQELHVSECAQRIKELSSA 1340
Query: 424 SQSAPLTVEEQVITIYTGTNGYLDSLEIRQVRKFLVELRAYLKTNKPQFNEIISSTKTFT 483
++ A T++ ++ TN ++ IR + L E A + K ++ TK +
Sbjct: 1341 AEKACATLK-----LFETTNNAAATV-IRDMENRLTE--ASVALEKAVLERDLNQTKVSS 1392
Query: 484 GEAEALLKEAIQEQMELFLLQEQVEK 509
EA+ E + + ++L L +V++
Sbjct: 1393 SEAKVESLEELCQDLKLQLENLRVKE 1418
>At4g18240 starch synthase-like protein
Length = 1071
Score = 30.4 bits (67), Expect = 2.4
Identities = 21/69 (30%), Positives = 31/69 (44%), Gaps = 10/69 (14%)
Query: 451 IRQVRKFLVELRAYLKTNKPQFNEIISSTKTFTGEAEAL----------LKEAIQEQMEL 500
IR K ++ L T N+I+S + GE L +K A QE+ +
Sbjct: 192 IRSAEKNILRLDEARATALDDLNKILSDKEALQGEINVLEMKLSETDERIKTAAQEKAHV 251
Query: 501 FLLQEQVEK 509
LL+EQ+EK
Sbjct: 252 ELLEEQLEK 260
>At1g04890 hypothetical protein
Length = 494
Score = 30.0 bits (66), Expect = 3.2
Identities = 24/93 (25%), Positives = 44/93 (46%), Gaps = 3/93 (3%)
Query: 417 LRELLKQSQSAPLTVEEQVITIYTGTNGYLDSLEIRQVRKFLVELRAYLKTNKPQFNEII 476
L ELLK+ ++A TV ++ + D E + L + +A ++ QF ++
Sbjct: 200 LEELLKEERAARATVCVELDKERSAAASAAD--EAMAMIHRLQDEKAAIEMEARQFQRLV 257
Query: 477 SSTKTFTGEAEALLKE-AIQEQMELFLLQEQVE 508
TF E +LK+ I+ + E L+++VE
Sbjct: 258 EERSTFDAEEMVILKDILIRREREKHFLEKEVE 290
>At5g60370 unknown protein (At5g60370)
Length = 413
Score = 29.3 bits (64), Expect = 5.4
Identities = 28/122 (22%), Positives = 57/122 (45%), Gaps = 16/122 (13%)
Query: 331 IPTNVISITDGQIFLSADLFNAGIRPAINVGISVSRVGSAAQIKAMKQVAGKLKLELAQF 390
IP ++S + I +A + I P++ +S SR+ + K ++ + K +L+
Sbjct: 42 IPIEIVSEEEMAILDAALAASRSILPSVIRSVSPSRITAGGSPKTIRSITLFSKRKLSAC 101
Query: 391 AELEAF---------AQFASDL------DKATQNQLARGQR-LRELLKQSQSAPLTVEEQ 434
+++E A +DL +K +N L G+R + + +K Q+ L +EE+
Sbjct: 102 SDIEESYLHRFRRNQALGVTDLTGTEWCEKQMENVLCFGRRKVNKAMKVGQARHLQLEEE 161
Query: 435 VI 436
V+
Sbjct: 162 VV 163
>At4g04320 malonyl-CoA decarboxylase like protein
Length = 517
Score = 28.9 bits (63), Expect = 7.0
Identities = 26/118 (22%), Positives = 52/118 (44%), Gaps = 6/118 (5%)
Query: 364 VSRVGSAAQIKAMKQVAGKLKLELAQFAELEAFAQFASDLDKATQNQLARGQ--RLRELL 421
+S+ A ++A + K L+QF E+++ Q D QN G R+ +++
Sbjct: 1 MSKKNLAILLRARMRSNNPSKFSLSQFPEIQSNPQENHSRDHIVQNSNDFGTTGRVYDVV 60
Query: 422 KQSQSAPLTVEEQ---VITIYTGTNGYLDSLEIRQVRKFLVELRAYLKTNKPQFNEII 476
+++ + ++ + IT+ GY SL + K L+ L N+ Q E++
Sbjct: 61 RETMHSAISASKTGVLDITLNDFQEGYF-SLSLEDREKLLLVLAKEYDVNREQVRELV 117
>At3g59010 pectinesterase precursor-like protein
Length = 529
Score = 28.9 bits (63), Expect = 7.0
Identities = 18/72 (25%), Positives = 34/72 (47%), Gaps = 5/72 (6%)
Query: 11 KIIRERIEQYNTEIKIV--NTGTVLQVGDGIARIYGLDEVMAGELVEFEEGTIGIALNLE 68
K++ +E+ + +GT + V + +A L++ ++ GT LN+
Sbjct: 216 KLLEASVEELRPHAVVAADGSGTHMSVAEALA---SLEKGSGRSVIHLTAGTYKENLNIP 272
Query: 69 SKNVGVVLMGDG 80
SK V+L+GDG
Sbjct: 273 SKQKNVMLVGDG 284
>At3g04340 unknown protein
Length = 1320
Score = 28.9 bits (63), Expect = 7.0
Identities = 41/160 (25%), Positives = 71/160 (43%), Gaps = 33/160 (20%)
Query: 167 LIIGDRQTGKTAVATD-------TILNQQGQNVICVYVAVGQKASSVAQVVNTLQERGAM 219
LI+G+R TGKT++A ++N + Q + + VGQ A++V ++ T ++ +
Sbjct: 821 LIVGERGTGKTSLALAIAAEARVPVVNVEAQE-LEAGLWVGQSAANVRELFQTARD---L 876
Query: 220 EYTIVVAETADSPATLQYLAPYTGAALAEFFMYRERHTLIIYDDLSKQ--------AQAY 271
I+ E D A ++ +T E F+ L+ D KQ + +
Sbjct: 877 APVIIFVEDFDLFAGVRGKFVHTKQQDHESFI---NQLLVELDGFEKQDGVVLMATTRNH 933
Query: 272 RQMSLLLRRPPGREAYPGDVFYLHS-------RLLERAAK 304
+Q+ LRRP + VF+L S R+L AA+
Sbjct: 934 KQIDEALRRPGRMDR----VFHLQSPTEMERERILHNAAE 969
Database: ara_mips
Posted date: Jul 15, 2004 10:29 AM
Number of letters in database: 2,978,382
Number of sequences in database: 6832
Database: /data/blast2/ara_mips_chr2
Posted date: Jul 15, 2004 10:29 AM
Number of letters in database: 1,737,135
Number of sequences in database: 4184
Database: /data/blast2/ara_mips_chr3
Posted date: Jul 15, 2004 10:29 AM
Number of letters in database: 2,236,886
Number of sequences in database: 5377
Database: /data/blast2/ara_mips_chr4
Posted date: Jul 15, 2004 10:29 AM
Number of letters in database: 1,748,816
Number of sequences in database: 4030
Database: /data/blast2/ara_mips_chr5
Posted date: Jul 15, 2004 10:29 AM
Number of letters in database: 2,569,679
Number of sequences in database: 6098
Database: /data/blast2/ara_mips_chl
Posted date: Jul 15, 2004 10:29 AM
Number of letters in database: 25,951
Number of sequences in database: 85
Database: /data/blast2/ara_mips_mit
Posted date: Jul 15, 2004 10:29 AM
Number of letters in database: 21,747
Number of sequences in database: 113
Lambda K H
0.316 0.134 0.355
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 9,348,510
Number of Sequences: 26719
Number of extensions: 371754
Number of successful extensions: 1131
Number of sequences better than 10.0: 24
Number of HSP's better than 10.0 without gapping: 12
Number of HSP's successfully gapped in prelim test: 12
Number of HSP's that attempted gapping in prelim test: 1102
Number of HSP's gapped (non-prelim): 28
length of query: 510
length of database: 11,318,596
effective HSP length: 104
effective length of query: 406
effective length of database: 8,539,820
effective search space: 3467166920
effective search space used: 3467166920
T: 11
A: 40
X1: 16 ( 7.3 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 41 (21.6 bits)
S2: 62 (28.5 bits)
Description of BAB33201.1