

BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
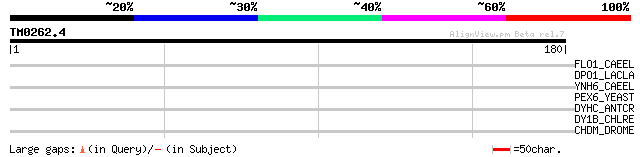
Query= TM0262.4
(180 letters)
Database: sprot
164,201 sequences; 59,974,054 total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
FLO1_CAEEL (Q17766) Folate-like transporter 1 31 1.8
DPO1_LACLA (Q9CDS1) DNA polymerase I (EC 2.7.7.7) (POL I) 31 1.8
YNH6_CAEEL (P32744) Hypothetical protein R107.6 in chromosome III 30 3.9
PEX6_YEAST (P33760) Peroxisomal biogenesis factor 6 (Peroxin-6) ... 30 3.9
DYHC_ANTCR (P39057) Dynein beta chain, ciliary 30 3.9
DY1B_CHLRE (Q9MBF8) Dynein 1-beta heavy chain, flagellar inner a... 29 6.7
CHDM_DROME (O97159) Chromodomain helicase-DNA-binding protein Mi... 28 8.8
>FLO1_CAEEL (Q17766) Folate-like transporter 1
Length = 360
Score = 30.8 bits (68), Expect = 1.8
Identities = 17/45 (37%), Positives = 28/45 (61%), Gaps = 5/45 (11%)
Query: 69 MAWFTTLPL----GSIVKFRDFSSKFLIQFSASKIKQVTIDDLYN 109
M+W TT + G++ +FR ++ FL F AS K +T+D+LY+
Sbjct: 1 MSWRTTTAILCLYGAVKEFRP-ATPFLTPFLASPEKNITLDELYS 44
>DPO1_LACLA (Q9CDS1) DNA polymerase I (EC 2.7.7.7) (POL I)
Length = 877
Score = 30.8 bits (68), Expect = 1.8
Identities = 16/58 (27%), Positives = 33/58 (56%), Gaps = 2/58 (3%)
Query: 77 LGSIVKFRDFSSKFLIQFSASKIKQVTIDDLYNVRQLERETLKQYVKRYSAASVKIEE 134
L + RDF SK L+Q I V +++L ++ L ++T+++ ++ A +K++E
Sbjct: 812 LDKALSARDFKSKLLLQVHDEIILDVPLEELDEIKVLVKQTMEEAIE--LAVPLKVDE 867
>YNH6_CAEEL (P32744) Hypothetical protein R107.6 in chromosome III
Length = 1063
Score = 29.6 bits (65), Expect = 3.9
Identities = 32/88 (36%), Positives = 42/88 (47%), Gaps = 11/88 (12%)
Query: 43 SMKMVISAASDAVKCRMFPSMFKGTAMAWFTTLPLGSIVKFRD---FSSKFLIQFSASKI 99
S K + ASDA + +GTA + + L GSI R+ SSKFL Q SAS I
Sbjct: 294 SSKQKMLRASDAASSSTSINSERGTA-PFRSKLSAGSIGGIRNAPNISSKFLAQRSASAI 352
Query: 100 --KQVT-----IDDLYNVRQLERETLKQ 120
KQVT + N+R + TL +
Sbjct: 353 DTKQVTRMATSVSRTPNIRPMTTRTLSK 380
>PEX6_YEAST (P33760) Peroxisomal biogenesis factor 6 (Peroxin-6)
(Peroxisome biosynthesis protein PAS8)
Length = 1030
Score = 29.6 bits (65), Expect = 3.9
Identities = 27/111 (24%), Positives = 45/111 (40%), Gaps = 19/111 (17%)
Query: 15 VAIPDNMKSLALEAYSGGTDPKDHLLYFSMKMVISAASDAVKCRMFPSMFKGTAMAWFTT 74
V+ DN+ +L +YS G P D +K ++ A R + K W
Sbjct: 647 VSYMDNISFSSLSSYSAGLTPLD------IKSIVETARMTATARFYQESKK---CGW--- 694
Query: 75 LPLGSIVKFRDFS-------SKFLIQFSASKIKQVTIDDLYNVRQLERETL 118
LP ++ D S ++F + A +I VT DD+ + ++ E L
Sbjct: 695 LPQSILITQEDLSKATSKARNEFSVSIGAPQIPNVTWDDIGGIDFVKGEIL 745
>DYHC_ANTCR (P39057) Dynein beta chain, ciliary
Length = 4466
Score = 29.6 bits (65), Expect = 3.9
Identities = 30/125 (24%), Positives = 58/125 (46%), Gaps = 15/125 (12%)
Query: 55 VKCRMFPSMFKGTAMAWFTTLPLGSIVKFRDFSSKFL--IQFSASKIKQVTIDDLYNVRQ 112
V+ R FP++ T++ WF P ++V S +FL ++ IK + + V
Sbjct: 2944 VRSRKFPAVVNCTSIDWFHEWPQEALV---SVSKRFLDEVELLKGDIKNSIAEFMAYVHV 3000
Query: 113 LERETLKQYV---KRYSAASVK--IEELEP*ACARAFKDGLLPGKLNNKLSRKLARSMTE 167
E+ KQY+ +RY+ + K +E+++ A K L K+ +L +T+
Sbjct: 3001 SVNESSKQYLTNERRYNYTTPKSFLEQIKLYESLLAMKSKELTAKM-----ERLENGLTK 3055
Query: 168 VRAQA 172
+++ A
Sbjct: 3056 LQSTA 3060
>DY1B_CHLRE (Q9MBF8) Dynein 1-beta heavy chain, flagellar inner arm I1
complex (1-beta DHC) (Dynein 1, subspecies f)
Length = 4513
Score = 28.9 bits (63), Expect = 6.7
Identities = 10/25 (40%), Positives = 15/25 (60%)
Query: 56 KCRMFPSMFKGTAMAWFTTLPLGSI 80
+CRMFP + T + WFT P ++
Sbjct: 2988 RCRMFPGLVNCTTIDWFTEWPADAL 3012
>CHDM_DROME (O97159) Chromodomain helicase-DNA-binding protein Mi-2
homolog (dMi-2)
Length = 1982
Score = 28.5 bits (62), Expect = 8.8
Identities = 13/36 (36%), Positives = 21/36 (58%)
Query: 49 SAASDAVKCRMFPSMFKGTAMAWFTTLPLGSIVKFR 84
+A++ A FP+ F+GT + FT+ P G+ FR
Sbjct: 1932 NASNAAQLMAQFPAGFQGTTLPAFTSGPAGNFANFR 1967
Database: sprot
Posted date: Nov 25, 2004 10:54 AM
Number of letters in database: 59,974,054
Number of sequences in database: 164,201
Lambda K H
0.323 0.135 0.381
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 18,076,761
Number of Sequences: 164201
Number of extensions: 621862
Number of successful extensions: 2035
Number of sequences better than 10.0: 7
Number of HSP's better than 10.0 without gapping: 3
Number of HSP's successfully gapped in prelim test: 4
Number of HSP's that attempted gapping in prelim test: 2031
Number of HSP's gapped (non-prelim): 8
length of query: 180
length of database: 59,974,054
effective HSP length: 103
effective length of query: 77
effective length of database: 43,061,351
effective search space: 3315724027
effective search space used: 3315724027
T: 11
A: 40
X1: 16 ( 7.5 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 40 (21.6 bits)
S2: 62 (28.5 bits)
Lotus: description of TM0262.4