

BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
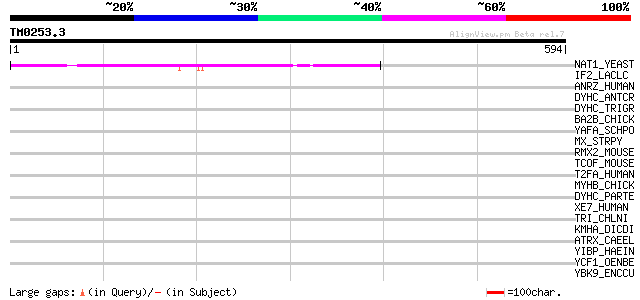
Query= TM0253.3
(594 letters)
Database: sprot
164,201 sequences; 59,974,054 total letters
Searching..................................................done
Score E
Sequences producing significant alignments: (bits) Value
NAT1_YEAST (P12945) N-terminal acetyltransferase 1 (EC 2.3.1.88)... 189 2e-47
IF2_LACLC (Q9X764) Translation initiation factor IF-2 41 0.010
ANRZ_HUMAN (Q6UB98) Ankyrin repeat domain protein 12 (Ankyrin re... 39 0.038
DYHC_ANTCR (P39057) Dynein beta chain, ciliary 39 0.050
DYHC_TRIGR (P23098) Dynein beta chain, ciliary 38 0.065
BA2B_CHICK (Q9DE13) Bromodomain adjacent to zinc finger domain 2... 38 0.065
YAFA_SCHPO (Q09863) Hypothetical protein C29E6.10c in chromosome I 38 0.085
MX_STRPY (P16946) Virulence factor-related M protein precursor 37 0.15
RMX2_MOUSE (Q8R0F5) RNA binding motif protein, X-linked 2 37 0.19
TCOF_MOUSE (O08784) Treacle protein (Treacher Collins syndrome p... 36 0.25
T2FA_HUMAN (P35269) Transcription initiation factor IIF, alpha s... 36 0.25
MYHB_CHICK (P10587) Myosin heavy chain, gizzard smooth muscle 36 0.25
DYHC_PARTE (Q27171) Dynein heavy chain, cytosolic (DYHC) 36 0.25
XE7_HUMAN (Q02040) B-lymphocyte antigen precursor (B-lymphocyte ... 36 0.32
TRI_CHLNI (Q7M3Y3) Troponin I (TnI) 36 0.32
KMHA_DICDI (P42527) Myosin heavy chain kinase A (EC 2.7.1.129) (... 36 0.32
ATRX_CAEEL (Q9U7E0) Transcriptional regulator ATRX homolog (X-li... 36 0.32
YIBP_HAEIN (P44864) Hypothetical protein HI0756 35 0.42
YCF1_OENBE (P31563) Hypothetical protein ycf1 (ORF 1005) (Fragment) 35 0.42
YBK9_ENCCU (Q8STZ1) Hypothetical protein ECU11_2090 35 0.42
>NAT1_YEAST (P12945) N-terminal acetyltransferase 1 (EC 2.3.1.88)
(Amino-terminal, alpha-amino, acetyltransferase 1)
Length = 853
Score = 189 bits (479), Expect = 2e-47
Identities = 130/441 (29%), Positives = 215/441 (48%), Gaps = 60/441 (13%)
Query: 2 IPLDFLQG-DRFREAADSYMRPLLTKGVPSLFSDLSSLYDHPGK--ANILEQLILELENS 58
IPL FLQ + + Y+ P L +GVP+ FS++ LY + +LE+++L+ +
Sbjct: 313 IPLTFLQDKEELSKKLREYVLPQLERGVPATFSNVKPLYQRRKSKVSPLLEKIVLDYLSG 372
Query: 59 IRTTGTYPGRVEKEPPSTLMWILFLLSQHYDRRSQYEIALSKINEAIEHTPTVIDLYSVK 118
+ T + P +W + LSQH+ + A I+ A++HTPT+++ Y +K
Sbjct: 373 LDPT---------QDPIPFIWTNYYLSQHFLFLKDFPKAQEYIDAALDHTPTLVEFYILK 423
Query: 119 SRILKHAGDLAAAAAFADEARCMDLSDRYVNSECVKRMLQADQVVLAEKTAVLFTKEGDQ 178
+RILKH G + AA +E R +DL DR++N + VK L+A+ + A + A LFTK D
Sbjct: 424 ARILKHLGLMDTAAGILEEGRQLDLQDRFINCKTVKYFLRANNIDKAVEVASLFTKNDDS 483
Query: 179 HN---NLHDMQCMWYELASAESYFR--------QGDL----------------------- 204
N +LH ++ W+ + AE+Y+R DL
Sbjct: 484 VNGIKDLHLVEASWFIVEQAEAYYRLYLDRKKKLDDLASLKKEVESDKSEQIANDIKENQ 543
Query: 205 -------GLALKKFLAVEKHYTDITEDQFDFHSYCLRKMTLRTYVEMLKFQDRLHSHVYF 257
GLALK+F A+ K Y +DQ DFHSYC+RK T R Y+EML++ L++ +
Sbjct: 544 WLVRKYKGLALKRFNAIPKFYKQFEDDQLDFHSYCMRKGTPRAYLEMLEWGKALYTKPMY 603
Query: 258 HKAAVGAIRCYIKLHDSPPKSTAEEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNE 317
+A A + Y ++HD K ++ D+ S + + + +++K AKKEA N+
Sbjct: 604 VRAMKEASKLYFQMHDDRLKRKSDSLDENSDEIQNNGQNSSSQKKK----AKKEAAAMNK 659
Query: 318 ESSASGISKSGKRQAKPVDPDPRGEKLLQVEDPLLE-ATKYLKLLQKNSPDSLETHLLSF 376
+KS D D GEKL++ P+ + AT++ + ++L F
Sbjct: 660 RKETE--AKSVAAYPSDQDNDVFGEKLIETSTPMEDFATEFYNNYSMQVREDERDYILDF 717
Query: 377 ELHMRRQKILLAFQAVKQLLR 397
E + R K+ L F ++ + +
Sbjct: 718 EFNYRIGKLALCFASLNKFAK 738
>IF2_LACLC (Q9X764) Translation initiation factor IF-2
Length = 950
Score = 40.8 bits (94), Expect = 0.010
Identities = 35/134 (26%), Positives = 62/134 (46%), Gaps = 10/134 (7%)
Query: 233 LRKMTLRTYVEMLKFQDRLHSHVYFHKAAVG--AIRCYIKLHDSPPKSTAEEDDDMSKLL 290
L K+ LR E K + H + KA V AI IK + K+ E + + +++
Sbjct: 65 LLKLKLRLVPETAKSKQEDHPRTFAGKAVVEDPAILARIKEKEEAKKAAKTEAEPIEEVI 124
Query: 291 PSQKKKLRQKQRKAEARAKKEAEE-KNEESSASGISKSGKRQAKPVDPDPRGEKLLQVED 349
++K K+ + +K+E +A +AEE K E+ A + + K + K E + ++
Sbjct: 125 TTEKPKVAEPVKKSEPKAAAKAEETKVEKVEAKANTVTPKAEVKT-------ENVADKKE 177
Query: 350 PLLEATKYLKLLQK 363
P++ K L QK
Sbjct: 178 PVVTEEKKKSLTQK 191
>ANRZ_HUMAN (Q6UB98) Ankyrin repeat domain protein 12 (Ankyrin
repeat-containing cofactor-2) (GAC-1 protein)
Length = 2062
Score = 38.9 bits (89), Expect = 0.038
Identities = 35/137 (25%), Positives = 66/137 (47%), Gaps = 14/137 (10%)
Query: 235 KMTLRTYVEMLKFQDRLHSHVYFHKAAVGAIRCYIKLHDSPPKSTAEEDDDMSKLLPSQ- 293
K+ L V+ +K +D+L+SH +C+ + S + ++ DD K
Sbjct: 857 KLDLSECVDKIKEKDKLYSH--------HTEKCHKEGEKSKNTAAIKKTDDREKSREKMD 908
Query: 294 ----KKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDPD-PRGEKLLQVE 348
K+K +++ AE++ K E+KN++S S SKS K + K D + + EK E
Sbjct: 909 RKHDKEKPEKERHLAESKEKHLMEKKNKQSDNSEYSKSEKGKNKEKDRELDKKEKSRDKE 968
Query: 349 DPLLEATKYLKLLQKNS 365
+ +K+++ +K+S
Sbjct: 969 SINITNSKHIQEEKKSS 985
>DYHC_ANTCR (P39057) Dynein beta chain, ciliary
Length = 4466
Score = 38.5 bits (88), Expect = 0.050
Identities = 31/106 (29%), Positives = 51/106 (47%), Gaps = 16/106 (15%)
Query: 277 KSTAEEDDDMSKLLPSQKKKLRQKQRKA-----------EARAKKEAEEKNEESSASGIS 325
+STA++ DD+ L SQ+ +L QK A E +K++A +EE + I+
Sbjct: 3057 QSTAQQVDDLKAKLASQEVELAQKNEDADKLIQVVGVETEKVSKEKATVDDEEKKVAIIN 3116
Query: 326 KSGKRQAKPVDPDPRGEKLLQVEDPLLEATKYLKLLQKNSPDSLET 371
+ ++AK D L + E LL A + L L KN+ L++
Sbjct: 3117 EEVSKKAKDCSED-----LAKAEPALLAAQEALNTLNKNNLTELKS 3157
>DYHC_TRIGR (P23098) Dynein beta chain, ciliary
Length = 4466
Score = 38.1 bits (87), Expect = 0.065
Identities = 31/106 (29%), Positives = 51/106 (47%), Gaps = 16/106 (15%)
Query: 277 KSTAEEDDDMSKLLPSQKKKLRQKQRKA-----------EARAKKEAEEKNEESSASGIS 325
+STA++ DD+ L SQ+ +L QK A E +K++A +EE + I+
Sbjct: 3057 QSTAQQVDDLKAKLASQEVELAQKNEDADKLIQVVGVETEKVSKEKAIADDEEKKVAIIN 3116
Query: 326 KSGKRQAKPVDPDPRGEKLLQVEDPLLEATKYLKLLQKNSPDSLET 371
+ ++AK D L + E LL A + L L KN+ L++
Sbjct: 3117 EEVSKKAKDCSED-----LAKAEPALLAAQEALNTLNKNNLTELKS 3157
>BA2B_CHICK (Q9DE13) Bromodomain adjacent to zinc finger domain 2B
(Extracellular matrix protein F22)
Length = 2130
Score = 38.1 bits (87), Expect = 0.065
Identities = 34/128 (26%), Positives = 63/128 (48%), Gaps = 9/128 (7%)
Query: 292 SQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDPDPRGEKLLQVE-DP 350
+Q K LR+ Q++ +ARA KEA +K + A+ + K Q K + + +++ Q+ +
Sbjct: 813 AQIKLLRKLQKQEQARAAKEA-KKQQAIMAAEEKRKQKEQIKIMKQQEKIKRIQQIRMEK 871
Query: 351 LLEATKYLKLLQKNSPDSLETHLLSFELHM------RRQKILLAFQAV-KQLLRLDAEHP 403
L A + L+ +K ++ LL E + R+Q +LL Q + + L ++ E
Sbjct: 872 ELRAQQILEAKKKKKEEAANAKLLEAEKRIKEKEMRRQQAVLLKHQELERHRLDMERERR 931
Query: 404 DSHRCLIK 411
H L+K
Sbjct: 932 RQHMMLMK 939
>YAFA_SCHPO (Q09863) Hypothetical protein C29E6.10c in chromosome I
Length = 1085
Score = 37.7 bits (86), Expect = 0.085
Identities = 40/162 (24%), Positives = 70/162 (42%), Gaps = 18/162 (11%)
Query: 282 EDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDPDPRG 341
E + + L QK++ +QKQ++ E + KK+ +E + E A + + + ++ R
Sbjct: 649 EQEKKQQELERQKREEKQKQKEREKKLKKQQQEADREKMAREQRLREEEEKRILEERKRR 708
Query: 342 EKLLQVEDPLLEATKYLKLLQKNSPDSLETHLLSFELHMRRQKILLAFQAVKQLLRLDAE 401
EKL + E E + +LL+K S + E +R KI F + D
Sbjct: 709 EKLDKEE----EERRRRELLEKESEEK--------ERRLREAKIAAFFAPNQTKEGSDGC 756
Query: 402 HPDSHRCLIKFFHKVGSLNTPVNDSEKLVWSVVEAERQTISQ 443
S + F K G L VND +KL ++++ + Q
Sbjct: 757 TTSSQ---LGLFEKKGDL---VNDEDKLSSHLLDSVPNALRQ 792
Score = 31.2 bits (69), Expect = 8.0
Identities = 39/208 (18%), Positives = 90/208 (42%), Gaps = 19/208 (9%)
Query: 215 EKHYTDITEDQFDFHSYCLRKMTLRTYVEMLK---FQDRLHSHVYFHKAAVGAIRCYIKL 271
E+ D+ ED+ D + R R ++ F+ R+ + ++ V R L
Sbjct: 517 EEDEEDVDEDELDLMTDEQRMEEGRRMFQIFAARLFEQRV---LQAYREKVAQQRQAKLL 573
Query: 272 HDSPPKSTAEEDDDMSKLLPSQKKKLRQKQ---RKAEARAKKEAEEKNEESSASGISKSG 328
+ ++ +++ ++ K+ +KK+ ++KQ K E R ++EAE E+++ +
Sbjct: 574 EEIEEENKRKQERELKKIREKEKKRDKKKQLKLAKEEERQRREAERLAEQAAQKALEAKR 633
Query: 329 KRQAKPVDPDPRGEKLLQVEDPLLEATK---------YLKLLQKNSPDSLETHLLSFELH 379
+ +A+ + R ++ + + LE K K L+K ++ + ++ E
Sbjct: 634 QEEARKKREEQRLKREQEKKQQELERQKREEKQKQKEREKKLKKQQQEA-DREKMAREQR 692
Query: 380 MRRQKILLAFQAVKQLLRLDAEHPDSHR 407
+R ++ + K+ +LD E + R
Sbjct: 693 LREEEEKRILEERKRREKLDKEEEERRR 720
>MX_STRPY (P16946) Virulence factor-related M protein precursor
Length = 369
Score = 37.0 bits (84), Expect = 0.15
Identities = 48/202 (23%), Positives = 88/202 (42%), Gaps = 22/202 (10%)
Query: 281 EEDDDMSKLLPSQKKKL---------------RQKQRKAEARAKKEAEEKNEESSASGIS 325
EE+ + SK L +K+KL ++K+ AK E + E+S G+S
Sbjct: 78 EEEQEKSKNLEKEKQKLENQALNFQDVIETQEKEKEDLKTTLAKATKENEISEASRKGLS 137
Query: 326 K--SGKRQAKPVDPDPRGEKLLQVEDPLLEATKYLKLLQKNSPDSLE-THLLSFELHMRR 382
+ R AK + + + +KL L EA + + +K + LE + EL +
Sbjct: 138 RDLEASRAAKK-ELEAKHQKLEAENKKLTEANQVSEASRKGLSNDLEASRAAKKELEAKY 196
Query: 383 QKILLAFQAVK-QLLRLDAEHPDSHRCLIKFFHKVGSLNTPVNDSEKLVWSVVEAERQTI 441
QK+ QA++ + +L+A+ P R K + L ++K+ + +A+ Q
Sbjct: 197 QKLETDHQALEAKHQKLEADLPKFQRPSRKGLSR--DLEASREANKKVTSELTQAKAQLS 254
Query: 442 SQLQGKSLFETNKSFLEKYEDS 463
+ + K L E K+ L+ D+
Sbjct: 255 ALEESKKLSEKEKAELQAKLDA 276
>RMX2_MOUSE (Q8R0F5) RNA binding motif protein, X-linked 2
Length = 326
Score = 36.6 bits (83), Expect = 0.19
Identities = 30/104 (28%), Positives = 47/104 (44%), Gaps = 21/104 (20%)
Query: 274 SPPKSTAEEDDDMSKLLPSQKK-KLRQKQRKAEARAKKE----------AEEKNEESSAS 322
SPP+ + +ED ++K KK K ++K+ K E +A+ E E+K+E +S
Sbjct: 144 SPPEVSEDEDAKLTKKHKKDKKEKKKRKKEKTEGQAQAEQPSCSRSATVKEKKDERASRK 203
Query: 323 GISKSGKRQAKPV----------DPDPRGEKLLQVEDPLLEATK 356
SK+ +R K PD R + EDP +A K
Sbjct: 204 HSSKTSERAQKSEHRESKKSHSGSPDGRSSYHARAEDPECKARK 247
>TCOF_MOUSE (O08784) Treacle protein (Treacher Collins syndrome
protein homolog)
Length = 1320
Score = 36.2 bits (82), Expect = 0.25
Identities = 20/59 (33%), Positives = 34/59 (56%), Gaps = 3/59 (5%)
Query: 279 TAEEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDP 337
+ E+ D SK S+KKK +K++ E + KK+ ++ + SAS I K K++ K +P
Sbjct: 1263 SGEQSDPKSK---SKKKKSLKKKKDKEKKEKKKGKKSLAKDSASPIQKKKKKKKKSAEP 1318
>T2FA_HUMAN (P35269) Transcription initiation factor IIF, alpha
subunit (EC 2.7.1.37) (TFIIF-alpha) (Transcription
initiation factor RAP74) (General transcription factor
IIF polypeptide 1 74 kDa subunit protein)
Length = 517
Score = 36.2 bits (82), Expect = 0.25
Identities = 22/72 (30%), Positives = 40/72 (55%), Gaps = 5/72 (6%)
Query: 273 DSPPKSTAEEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKS---GK 329
+ PP+ EE+++ K P+ ++K R+K E+ + +E++ +E SSA ++K K
Sbjct: 314 EKPPEEDKEEEEE--KKAPTPQEKKRRKDSSEESDSSEESDIDSEASSALFMAKKKTPPK 371
Query: 330 RQAKPVDPDPRG 341
R+ KP RG
Sbjct: 372 RERKPSGGSSRG 383
>MYHB_CHICK (P10587) Myosin heavy chain, gizzard smooth muscle
Length = 1978
Score = 36.2 bits (82), Expect = 0.25
Identities = 57/258 (22%), Positives = 111/258 (42%), Gaps = 33/258 (12%)
Query: 281 EEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDPDPR 340
EE+++ S+ L ++KKK++Q+ E + ++E E ++ ++ GK K ++ D
Sbjct: 937 EEEEERSQQLQAEKKKMQQQMLDLEEQLEEE-EAARQKLQLEKVTADGK--IKKMEDD-- 991
Query: 341 GEKLLQVEDPLLEATKYLKLLQKNSPDSLETHL-------------------LSFELHMR 381
+L +ED + TK KLL++ D L T+L + EL +R
Sbjct: 992 ---ILIMEDQNNKLTKERKLLEERVSD-LTTNLAEEEEKAKNLTKLKNKHESMISELEVR 1047
Query: 382 RQKILLAFQAVKQLLR-LDAEHPDSHRCLIKFFHKVGSLNTPVNDSEKLVWSVVEAERQT 440
+K + Q ++++ R L+ E D H + + ++ L + E+ + + +
Sbjct: 1048 LKKEEKSRQELEKIKRKLEGESSDLHEQIAELQAQIAELKAQLAKKEEELQAALARLEDE 1107
Query: 441 ISQLQG--KSLFETNKSFLEKYEDSLMHRAAFGEM-MYVLDPSRRSEAVKI-IEGSTNSP 496
SQ K + E + ED +AA + D S EA+K +E + ++
Sbjct: 1108 TSQKNNALKKIRELESHISDLQEDLESEKAARNKAEKQKRDLSEELEALKTELEDTLDTT 1167
Query: 497 VSRNGTNWKLEDCIEVHK 514
++ K E + V K
Sbjct: 1168 ATQQELRAKREQEVTVLK 1185
>DYHC_PARTE (Q27171) Dynein heavy chain, cytosolic (DYHC)
Length = 4540
Score = 36.2 bits (82), Expect = 0.25
Identities = 28/107 (26%), Positives = 57/107 (53%), Gaps = 11/107 (10%)
Query: 236 MTLRTYVEMLKFQDRLHSHVYFHKAAVGAIRCYIKLHDSPPKSTAEEDDDMSKLLPSQKK 295
+T R Y++ LK ++LH+ K+ + + ++ + K T ++ +M K L +K
Sbjct: 3059 ITPRDYLDFLKHFEKLHNE---KKSQLEDQQLHLNVGLDKLKETEQQVLEMQKSLDQKKV 3115
Query: 296 KLRQKQRKAEAR------AKKEAEEKNEESSASGISKSGKRQAKPVD 336
+L K+R+A + KK AE+K E+S+ +S +++AK ++
Sbjct: 3116 ELLTKERQAGEKLQTIIEEKKIAEKKKEDSTR--LSSDAEKKAKEME 3160
>XE7_HUMAN (Q02040) B-lymphocyte antigen precursor (B-lymphocyte
surface antigen) (721P) (Protein XE7)
Length = 695
Score = 35.8 bits (81), Expect = 0.32
Identities = 42/169 (24%), Positives = 74/169 (42%), Gaps = 18/169 (10%)
Query: 239 RTYVEMLKFQDRLHSHVYFHKAAVG-AIRCYIKLHDSPPK-----STAEEDDDMSKLLPS 292
R Y+ ++ L +K G A+ C IK+ K S + + KL
Sbjct: 222 REYMGFIQAMSALRGMKLMYKGEDGKAVACNIKVSFDSTKHLSDASIKKRQLERQKLQEL 281
Query: 293 QKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDPDPRGEKLLQVEDPLL 352
++++ QK+R+ EA ++ AEE+ ++ + + KR+ K R + Q + L
Sbjct: 282 EQQREEQKRREKEAEERQRAEER-KQKELEELERERKREEK-----LRKREQKQRDRELR 335
Query: 353 EATKYLKLLQKNSPDSLETHLLSFELHMRRQKILLAFQAVKQLLRLDAE 401
K L+ LQ L+ ++ + +K+LLA Q Q +RL AE
Sbjct: 336 RNQKKLEKLQAEEQKQLQE-----KIKLEERKLLLA-QRNLQSIRLIAE 378
>TRI_CHLNI (Q7M3Y3) Troponin I (TnI)
Length = 292
Score = 35.8 bits (81), Expect = 0.32
Identities = 45/174 (25%), Positives = 73/174 (41%), Gaps = 14/174 (8%)
Query: 299 QKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDPDPRGEKLLQVEDPLLEA-TKY 357
+++R+ EAR E ++K ++ G+S K+ K + E L EA KY
Sbjct: 119 EQEREEEARRMAEEQKKKKKKGLGGLSPEKKKMLKKLIMQKAAEDLANEAKAKAEAKEKY 178
Query: 358 LK-LLQKNSPDSLETHLLSFELHMRRQKILLAFQAVKQLLRLDAEHPDSHRCLIKFFHKV 416
+ L+ K S D + L Q + F K+L L+ + D + K ++
Sbjct: 179 INDLVPKFSTDGKDVAAL--------QALCKDFH--KRLASLEEDVYDWEAKIRKQDFEI 228
Query: 417 GSLNTPVNDSE-KLVWSVVEAERQTISQLQGKSLFETNKS-FLEKYEDSLMHRA 468
L VND++ K V V+ +T S+L E KS F + + S H A
Sbjct: 229 NELTLKVNDTKGKFVKPVLRKVNKTESKLDKIQRKEAKKSDFRDNLKSSREHEA 282
>KMHA_DICDI (P42527) Myosin heavy chain kinase A (EC 2.7.1.129)
(MHCK A)
Length = 1146
Score = 35.8 bits (81), Expect = 0.32
Identities = 24/82 (29%), Positives = 40/82 (48%), Gaps = 6/82 (7%)
Query: 284 DDMSKLLPSQKKKLRQKQRKAEARAKK-EAEEKNEESSASGISKSGKRQAKPVDPDPRGE 342
+++ K+ +KK+R++ +A K E K+ S G+ K K Q D +
Sbjct: 369 EEVKKVEEKLQKKIREEIDNTKAELSKVERSVKDNRSEIEGLEKDCKNQF-----DKQDN 423
Query: 343 KLLQVEDPLLEATKYLKLLQKN 364
K+ QVED L ++ L L+Q N
Sbjct: 424 KIKQVEDDLKKSDSLLLLMQNN 445
>ATRX_CAEEL (Q9U7E0) Transcriptional regulator ATRX homolog
(X-linked nuclear protein-1)
Length = 1359
Score = 35.8 bits (81), Expect = 0.32
Identities = 16/65 (24%), Positives = 35/65 (53%)
Query: 279 TAEEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDPD 338
++EE ++ K+ S+K K + +++AE + + +EK + S G+ K K +++ D
Sbjct: 155 SSEESEEERKVKKSKKNKEKSVKKRAETSEESDEDEKPSKKSKKGLKKKAKSESESESED 214
Query: 339 PRGEK 343
+ K
Sbjct: 215 EKEVK 219
Score = 34.7 bits (78), Expect = 0.72
Identities = 23/91 (25%), Positives = 44/91 (48%), Gaps = 6/91 (6%)
Query: 278 STAEEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDP 337
S+ +ED D + S+KK + K++ + +++ EE+ + S KS K++A+ +
Sbjct: 126 SSEDEDSDEEREQKSKKKSKKTKKQTSSESSEESEEERKVKKSKKNKEKSVKKRAETSEE 185
Query: 338 DPRGEKLLQVEDPLLEATKYLKLLQKNSPDS 368
EK P ++ K LK K+ +S
Sbjct: 186 SDEDEK------PSKKSKKGLKKKAKSESES 210
Score = 33.9 bits (76), Expect = 1.2
Identities = 17/56 (30%), Positives = 32/56 (56%)
Query: 278 STAEEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAK 333
S++EEDDD + P + K +K+ K+E+ + + EE++ + S S K++ K
Sbjct: 62 SSSEEDDDDEEESPRKSSKKSRKRAKSESESDESDEEEDRKKSKSKKKVDQKKKEK 117
Score = 32.3 bits (72), Expect = 3.6
Identities = 27/93 (29%), Positives = 49/93 (52%), Gaps = 13/93 (13%)
Query: 279 TAEEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDPD 338
T+EE D+ K PS+K K + ++KA++ ++ E+E++ E + KS K+ K V +
Sbjct: 182 TSEESDEDEK--PSKKSK-KGLKKKAKSESESESEDEKE------VKKSKKKSKKVVKKE 232
Query: 339 PRGEKLLQVEDPLLEATKYLKLLQKNSPDSLET 371
E E P + T+ K + +S +S E+
Sbjct: 233 SESED----EAPEKKKTEKRKRSKTSSEESSES 261
Score = 31.2 bits (69), Expect = 8.0
Identities = 21/94 (22%), Positives = 45/94 (47%), Gaps = 1/94 (1%)
Query: 277 KSTAEEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVD 336
K T+ E + S+ KK + K++ + RA+ +EE +E+ S SK G ++ +
Sbjct: 149 KQTSSESSEESEEERKVKKSKKNKEKSVKKRAET-SEESDEDEKPSKKSKKGLKKKAKSE 207
Query: 337 PDPRGEKLLQVEDPLLEATKYLKLLQKNSPDSLE 370
+ E +V+ ++ K +K ++ ++ E
Sbjct: 208 SESESEDEKEVKKSKKKSKKVVKKESESEDEAPE 241
>YIBP_HAEIN (P44864) Hypothetical protein HI0756
Length = 410
Score = 35.4 bits (80), Expect = 0.42
Identities = 24/78 (30%), Positives = 38/78 (47%), Gaps = 1/78 (1%)
Query: 283 DDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAKPVDPDPRGE 342
D D L + ++ LRQ+ ++AE +A +E E++ E+ A KR +KP P +
Sbjct: 216 DQDKLNTLKANEQALRQEIQRAE-QAAREQEKREREALAQRQKAEEKRTSKPYQPTVQER 274
Query: 343 KLLQVEDPLLEATKYLKL 360
+LL L A K L
Sbjct: 275 QLLNSTSGLGAAKKQYSL 292
>YCF1_OENBE (P31563) Hypothetical protein ycf1 (ORF 1005) (Fragment)
Length = 1005
Score = 35.4 bits (80), Expect = 0.42
Identities = 18/57 (31%), Positives = 32/57 (55%)
Query: 277 KSTAEEDDDMSKLLPSQKKKLRQKQRKAEARAKKEAEEKNEESSASGISKSGKRQAK 333
KS E K + S++KK++ KQ+K +++ KK ++NE S KS +++ K
Sbjct: 774 KSKQNEIKSKQKKVKSKQKKVKSKQKKVKSKQKKVKSKQNEIKSKQNEIKSKQKKVK 830
>YBK9_ENCCU (Q8STZ1) Hypothetical protein ECU11_2090
Length = 634
Score = 35.4 bits (80), Expect = 0.42
Identities = 25/79 (31%), Positives = 41/79 (51%), Gaps = 6/79 (7%)
Query: 281 EEDDDMSKLLPSQKKKLRQKQRKAEARAKKE-----AEEKNEESSASGISKSGKRQAKPV 335
E + +LL +++ R+K+ +++ R KK+ A E EES G K G + A
Sbjct: 340 ERAKNEEELLRMVEREEREKREESKGRGKKKRGNRGAGESKEESKGRGKRKRGNKGAGES 399
Query: 336 DPDPRGEK-LLQVEDPLLE 353
+ RGE+ ++ EDPL E
Sbjct: 400 KEEDRGEEGGVEAEDPLEE 418
Database: sprot
Posted date: Nov 25, 2004 10:54 AM
Number of letters in database: 59,974,054
Number of sequences in database: 164,201
Lambda K H
0.317 0.132 0.378
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 68,773,306
Number of Sequences: 164201
Number of extensions: 2885318
Number of successful extensions: 13412
Number of sequences better than 10.0: 146
Number of HSP's better than 10.0 without gapping: 36
Number of HSP's successfully gapped in prelim test: 118
Number of HSP's that attempted gapping in prelim test: 12739
Number of HSP's gapped (non-prelim): 519
length of query: 594
length of database: 59,974,054
effective HSP length: 116
effective length of query: 478
effective length of database: 40,926,738
effective search space: 19562980764
effective search space used: 19562980764
T: 11
A: 40
X1: 16 ( 7.3 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 41 (21.7 bits)
S2: 69 (31.2 bits)
Lotus: description of TM0253.3