

BLAST2 result
BLASTP 2.2.2 [Dec-14-2001]
Reference: Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schaffer,
Jinghui Zhang, Zheng Zhang, Webb Miller, and David J. Lipman (1997),
"Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs", Nucleic Acids Res. 25:3389-3402.
Query= TM0134.10
(1564 letters)
Database: sprot
164,201 sequences; 59,974,054 total letters
Searching..................................................done
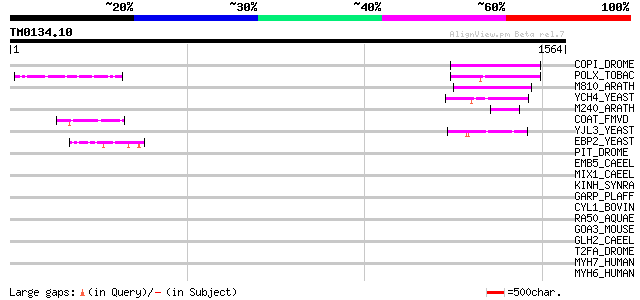
Score E
Sequences producing significant alignments: (bits) Value
COPI_DROME (P04146) Copia protein (Gag-int-pol protein) [Contain... 153 4e-36
POLX_TOBAC (P10978) Retrovirus-related Pol polyprotein from tran... 149 8e-35
M810_ARATH (P92519) Hypothetical mitochondrial protein AtMg00810... 145 6e-34
YCH4_YEAST (P25600) Transposon Ty5-1 34.5 kDa hypothetical protein 89 7e-17
M240_ARATH (P93290) Hypothetical mitochondrial protein AtMg00240... 63 6e-09
COAT_FMVD (P09519) Probable coat protein 59 1e-07
YJL3_YEAST (P47024) Transposon Ty4 207.7 kDa hypothetical protein 48 2e-04
EBP2_YEAST (P36049) rRNA processing protein EBP2 (EBNA1-binding ... 47 5e-04
PIT_DROME (Q9VD51) Probable ATP-dependent helicase pitchoune 44 0.005
EMB5_CAEEL (P34703) Abnormal embryogenesis protein 5 44 0.005
MIX1_CAEEL (Q09591) Mitotic chromosome and X-chromosome associat... 43 0.008
KINH_SYNRA (O43093) Kinesin heavy chain (Synkin) 43 0.008
GARP_PLAFF (P13816) Glutamic acid-rich protein precursor 42 0.017
CYL1_BOVIN (P35662) Cylicin I (Multiple-band polypeptide I) 42 0.017
RA50_AQUAE (O67124) Probable DNA double-strand break repair rad5... 41 0.023
GOA3_MOUSE (P55937) Golgi autoantigen, golgin subfamily A member... 41 0.023
GLH2_CAEEL (Q966L9) ATP-dependent RNA helicase glh-2 (EC 3.6.1.-... 41 0.029
T2FA_DROME (Q05913) Transcription initiation factor IIF, alpha s... 40 0.038
MYH7_HUMAN (P12883) Myosin heavy chain, cardiac muscle beta isof... 40 0.038
MYH6_HUMAN (P13533) Myosin heavy chain, cardiac muscle alpha iso... 40 0.038
>COPI_DROME (P04146) Copia protein (Gag-int-pol protein) [Contains:
Copia VLP protein; Copia protease (EC 3.4.23.-)]
Length = 1409
Score = 153 bits (386), Expect = 4e-36
Identities = 88/259 (33%), Positives = 146/259 (55%), Gaps = 6/259 (2%)
Query: 1243 NDILIVQIYVDDIIFGSANQSLCKEFSEMMQAEFEMSMMGELKYFLGIQVDQTPEGTYIH 1302
N+ + V +YVDD++ + + + F + +F M+ + E+K+F+GI+++ + Y+
Sbjct: 1079 NENIYVLLYVDDVVIATGDMTRMNNFKRYLMEKFRMTDLNEIKHFIGIRIEMQEDKIYLS 1138
Query: 1303 QSKYTKELLKKFNMLESTVAKTPMHPTCILEKEDASGKVCQKLYHGMIGTLLY-LTASRP 1361
QS Y K++L KFNM TP+ P+ I + S + C +IG L+Y + +RP
Sbjct: 1139 QSAYVKKILSKFNMENCNAVSTPL-PSKINYELLNSDEDCNTPCRSLIGCLMYIMLCTRP 1197
Query: 1362 DILFSVHLCARFQSDPRETHLTAVKRILRYLKGTTNLGLMYKK--T*EYKLSGYCDADYA 1419
D+ +V++ +R+ S +KR+LRYLKGT ++ L++KK E K+ GY D+D+A
Sbjct: 1198 DLTTAVNILSRYSSKNNSELWQNLKRVLRYLKGTIDMKLIFKKNLAFENKIIGYVDSDWA 1257
Query: 1420 GDRTERKSTSENC-QFLGSNLVSWASKWQSTIALSTAEAEYILTAICSTQMLWMKHQLED 1478
G +RKST+ + NL+ W +K Q+++A S+ EAEY+ + LW+K L
Sbjct: 1258 GSEIDRKSTTGYLFKMFDFNLICWNTKRQNSVAASSTEAEYMALFEAVREALWLKFLLTS 1317
Query: 1479 YQI-LESNISIYCDNTAAI 1496
I LE+ I IY DN I
Sbjct: 1318 INIKLENPIKIYEDNQGCI 1336
Score = 42.4 bits (98), Expect = 0.010
Identities = 58/262 (22%), Positives = 103/262 (39%), Gaps = 20/262 (7%)
Query: 50 ARAILLSAISYEEYEKITDREYAKGIFESLKMSHEGNKKVKESKALSLIQKYESFIMEPN 109
A++ ++ +S T A+ I E+L +E + L+L ++ S +
Sbjct: 55 AKSTIIEYLSDSFLNFATSDITARQILENLDAVYERKSLASQ---LALRKRLLSLKLSSE 111
Query: 110 ESIEEMFSRFQLLVAGIRPLNKSYTTKYHVIRVIRSLPESWMPLVTSIELTRDVERMSLE 169
S+ F F L++ + + ++ +LP + ++T+IE T E ++L
Sbjct: 112 MSLLSHFHIFDELISELLAAGAKIEEMDKISHLLITLPSCYDGIITAIE-TLSEENLTLA 170
Query: 170 ELISILKCHELK-HSEMLDSDEDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKK 228
+ + L E+K ++ D+ + + I N +K+ K R + K K G K
Sbjct: 171 FVKNRLLDQEIKIKNDHNDTSKKVMNAIVHNNNNTYKNNLFKNRVT---KPKKIFKGNSK 227
Query: 229 SSIKEVTCFECKESGHYKSDCPKLK-----KDKRPKKHFKTKKSLMVTFDESESEDVDYD 283
+K C C GH K DC K K+K +K +T S + F E +
Sbjct: 228 YKVK---CHHCGREGHIKKDCFHYKRILNNKNKENEKQVQTATSHGIAFMVKEVNNT--- 281
Query: 284 GEVQGLMDIVKDKGAESKDVVD 305
V V D GA + D
Sbjct: 282 -SVMDNCGFVLDSGASDHLIND 302
>POLX_TOBAC (P10978) Retrovirus-related Pol polyprotein from
transposon TNT 1-94 [Contains: Protease (EC 3.4.23.-);
Reverse transcriptase (EC 2.7.7.49); Endonuclease]
Length = 1328
Score = 149 bits (375), Expect = 8e-35
Identities = 88/268 (32%), Positives = 145/268 (53%), Gaps = 18/268 (6%)
Query: 1242 QNDILIVQIYVDDIIFGSANQSLCKEFSEMMQAEFEMSMMGELKYFLGIQV--DQTPEGT 1299
+N+ +I+ +YVDD++ ++ L + + F+M +G + LG+++ ++T
Sbjct: 999 ENNFIILLLYVDDMLIVGKDKGLIAKLKGDLSKSFDMKDLGPAQQILGMKIVRERTSRKL 1058
Query: 1300 YIHQSKYTKELLKKFNMLESTVAKTP----------MHPTCILEKEDASGKVCQKLYHGM 1349
++ Q KY + +L++FNM + TP M PT + EK G + + Y
Sbjct: 1059 WLSQEKYIERVLERFNMKNAKPVSTPLAGHLKLSKKMCPTTVEEK----GNMAKVPYSSA 1114
Query: 1350 IGTLLY-LTASRPDILFSVHLCARFQSDPRETHLTAVKRILRYLKGTTNLGLMYKKT*EY 1408
+G+L+Y + +RPDI +V + +RF +P + H AVK ILRYL+GTT L + + +
Sbjct: 1115 VGSLMYAMVCTRPDIAHAVGVVSRFLENPGKEHWEAVKWILRYLRGTTGDCLCFGGS-DP 1173
Query: 1409 KLSGYCDADYAGDRTERKSTSENCQFLGSNLVSWASKWQSTIALSTAEAEYILTAICSTQ 1468
L GY DAD AGD RKS++ +SW SK Q +ALST EAEYI +
Sbjct: 1174 ILKGYTDADMAGDIDNRKSSTGYLFTFSGGAISWQSKLQKCVALSTTEAEYIAATETGKE 1233
Query: 1469 MLWMKHQLEDYQILESNISIYCDNTAAI 1496
M+W+K L++ + + +YCD+ +AI
Sbjct: 1234 MIWLKRFLQELGLHQKEYVVYCDSQSAI 1261
Score = 60.8 bits (146), Expect = 3e-08
Identities = 73/306 (23%), Positives = 130/306 (41%), Gaps = 27/306 (8%)
Query: 15 IIVDGYERPVDEEGKKIPRSEMTADQKKLYSQHHKARAILLSAISYEEYEKITDREYAKG 74
+I G + +D + KK P + D L +A + + +S + I D + A+G
Sbjct: 28 LIQQGLHKVLDVDSKK-PDTMKAEDWADL---DERAASAIRLHLSDDVVNNIIDEDTARG 83
Query: 75 IF---ESLKMSHEGNKKVKESKALSLIQKYESFIMEPNESIEEMFSRFQLLVAGIRPLNK 131
I+ ESL MS K+ K L + M + + F L+ + L
Sbjct: 84 IWTRLESLYMSKTLTNKLYLKKQLYALH------MSEGTNFLSHLNVFNGLITQLANLGV 137
Query: 132 SYTTKYHVIRVIRSLPESWMPLVTSIELTRDVERMSLEELISILKCHELKHSEMLDSDED 191
+ I ++ SLP S+ L T+I + L+++ S L +E + + +
Sbjct: 138 KIEEEDKAILLLNSLPSSYDNLATTI--LHGKTTIELKDVTSALLLNEKMRKKPENQGQ- 194
Query: 192 ELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKKSSIKEVTCFECKESGHYKSDCPK 251
LI++ R ++ + Y SG A+GKS++ + KS ++ C+ C + GH+K DCP
Sbjct: 195 --ALITEGRGRSYQRSSNNYGRSG-ARGKSKN--RSKSRVRN--CYNCNQPGHFKRDCPN 247
Query: 252 LKKDKRPKKHFKTKKSLMVTFDESESEDVDYDGEVQGLMDIVKDKGAESKDVVDSDSESE 311
+K K K + +++ V + E + M + G ES+ VVD+ +
Sbjct: 248 PRKGKGETSGQKNDDNTAAMVQNNDNV-VLFINEEEECMHL---SGPESEWVVDTAASHH 303
Query: 312 GDPNSD 317
P D
Sbjct: 304 ATPVRD 309
>M810_ARATH (P92519) Hypothetical mitochondrial protein AtMg00810
(ORF240b)
Length = 240
Score = 145 bits (367), Expect = 6e-34
Identities = 72/222 (32%), Positives = 124/222 (55%), Gaps = 1/222 (0%)
Query: 1250 IYVDDIIFGSANQSLCKEFSEMMQAEFEMSMMGELKYFLGIQVDQTPEGTYIHQSKYTKE 1309
+YVDDI+ ++ +L + + F M +G + YFLGIQ+ P G ++ Q+KY ++
Sbjct: 5 LYVDDILLTGSSNTLLNMLIFQLSSTFSMKDLGPVHYFLGIQIKTHPSGLFLSQTKYAEQ 64
Query: 1310 LLKKFNMLESTVAKTPMHPTCILEKEDASGKVCQKLYHGMIGTLLYLTASRPDILFSVHL 1369
+L ML+ TP+ P + + + ++G L YLT +RPDI ++V++
Sbjct: 65 ILNNAGMLDCKPMSTPL-PLKLNSSVSTAKYPDPSDFRSIVGALQYLTLTRPDISYAVNI 123
Query: 1370 CARFQSDPRETHLTAVKRILRYLKGTTNLGLMYKKT*EYKLSGYCDADYAGDRTERKSTS 1429
+ +P +KR+LRY+KGT GL K + + +CD+D+AG + R+ST+
Sbjct: 124 VCQRMHEPTLADFDLLKRVLRYVKGTIFHGLYIHKNSKLNVQAFCDSDWAGCTSTRRSTT 183
Query: 1430 ENCQFLGSNLVSWASKWQSTIALSTAEAEYILTAICSTQMLW 1471
C FLG N++SW++K Q T++ S+ E EY A+ + ++ W
Sbjct: 184 GFCTFLGCNIISWSAKRQPTVSRSSTETEYRALALTAAELTW 225
>YCH4_YEAST (P25600) Transposon Ty5-1 34.5 kDa hypothetical protein
Length = 308
Score = 89.4 bits (220), Expect = 7e-17
Identities = 61/243 (25%), Positives = 114/243 (46%), Gaps = 17/243 (6%)
Query: 1228 CKG*SRYNSLLKNIQNDILIVQIYVDDIIFGSANQSLCKEFSEMMQAEFEMSMMGELKYF 1287
C+ + ++ + + + +YVDD++ + + + + + + M +G++ F
Sbjct: 64 CRHEGEHGLYFRSTSDGPIYIGVYVDDLLVAAPSPKIYDRVKQELTKLYSMKDLGKVDKF 123
Query: 1288 LGIQVDQTPEGT-------YIHQSKYTKELLKKFNMLESTVAKT-PMHPTCILEKEDASG 1339
LG+ + Q+ G YI ++ E+ F + ++ + + P+ T +D +
Sbjct: 124 LGLNIHQSTNGDITLSLQDYIAKAASESEI-NTFKLTQTPLCNSKPLFETTSPHLKDITP 182
Query: 1340 KVCQKLYHGMIGTLLYLT-ASRPDILFSVHLCARFQSDPRETHLTAVKRILRYLKGTTNL 1398
Y ++G LL+ RPDI + V L +RF +PR HL + +R+LRYL T ++
Sbjct: 183 ------YQSIVGQLLFCANTGRPDISYPVSLLSRFLREPRAIHLESARRVLRYLYTTRSM 236
Query: 1399 GLMYKKT*EYKLSGYCDADYAGDRTERKSTSENCQFLGSNLVSWAS-KWQSTIALSTAEA 1457
L Y+ + L+ YCDA + ST L V+W+S K + I + + EA
Sbjct: 237 CLKYRSGSQVALTVYCDASHGAIHDLPHSTGGYVTLLAGAPVTWSSKKLKGVIPVPSTEA 296
Query: 1458 EYI 1460
EYI
Sbjct: 297 EYI 299
>M240_ARATH (P93290) Hypothetical mitochondrial protein AtMg00240
(ORF111a)
Length = 111
Score = 63.2 bits (152), Expect = 6e-09
Identities = 29/82 (35%), Positives = 48/82 (58%)
Query: 1354 LYLTASRPDILFSVHLCARFQSDPRETHLTAVKRILRYLKGTTNLGLMYKKT*EYKLSGY 1413
+YLT +RPD+ F+V+ ++F S R + AV ++L Y+KGT GL Y T + +L +
Sbjct: 1 MYLTITRPDLTFAVNRLSQFSSASRTAQMQAVYKVLHYVKGTVGQGLFYSATSDLQLKAF 60
Query: 1414 CDADYAGDRTERKSTSENCQFL 1435
D+D+A R+S + C +
Sbjct: 61 ADSDWASCPDTRRSVTGFCSLV 82
>COAT_FMVD (P09519) Probable coat protein
Length = 489
Score = 58.9 bits (141), Expect = 1e-07
Identities = 55/204 (26%), Positives = 88/204 (42%), Gaps = 25/204 (12%)
Query: 131 KSYTTKYHVIRVIRSLPESWMPLVTSIELTRDVERMS--------LEELISILKCHELKH 182
KS+TTKY + R + E ++ ++ S L+ L+S C + K
Sbjct: 300 KSFTTKYSLAFAKRIIQEEIAKYCDFQRTSKKLKNFSKKCCSKNSLDPLVSF-GCRDTKK 358
Query: 183 SEMLDSDEDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKKSSIKEVTCFECKES 242
+ S + + K L ++WK K+ K+ GK K + K+ C+ C E
Sbjct: 359 KDFKKSSKYKAYKKKKTLKKLWKKKKRKFT-PGKYFSKKKPEKFCPQGRKKCRCWICTEE 417
Query: 243 GHYKSDCPKLKKDKRPKKHFKTKKSLMVTFDES--ESEDVDYDGEVQGL-MDIVKDKGAE 299
GHY ++CP K H + K L+ +E ED Y G ++ M+I+++ +E
Sbjct: 418 GHYANECP------NRKSHQEKVKILIHGMNEGYYPLEDA-YTGNLEVFSMEIIEETTSE 470
Query: 300 SKDVVDSDSESEGDPNSDDENEVF 323
+ DSDS S SDDE F
Sbjct: 471 EESTTDSDSSS-----SDDEQLSF 489
>YJL3_YEAST (P47024) Transposon Ty4 207.7 kDa hypothetical protein
Length = 1803
Score = 48.1 bits (113), Expect = 2e-04
Identities = 57/247 (23%), Positives = 101/247 (40%), Gaps = 25/247 (10%)
Query: 1234 YNSLLKNIQNDILIVQIYVDDIIFGSANQSLCKEFSEMMQAEFEMSMMGEL------KYF 1287
Y L ++ L++ +YVDD + ++N+ EF +++ FE+ + G L
Sbjct: 1444 YTPGLYQTEDKNLMIAVYVDDCVIAASNEQRLDEFINKLKSNFELKITGTLIDDVLDTDI 1503
Query: 1288 LGIQ---------VDQTPEGTYIHQSKYTKELLKKFNMLE----STVAKTPMHPTCILEK 1334
LG+ +D T + K E LKK ST P + +
Sbjct: 1504 LGMDLVYNKRLGTIDLTLKSFINRMDKKYNEELKKIRKSSIPHMSTYKIDPKKDVLQMSE 1563
Query: 1335 EDASGKVCQKLYHGMIGTLLYLT-ASRPDILFSVHLCARFQSDPRETHLTAVKRILRYLK 1393
E+ V + ++G L Y+ R DI F+V AR + P E + +I++YL
Sbjct: 1564 EEFRQGVLK--LQQLLGELNYVRHKCRYDIEFAVKKVARLVNYPHERVFYMIYKIIQYLV 1621
Query: 1394 GTTNLGLMYKK--T*EYKLSGYCDADYAGDRTERKSTSENCQFLGSNLVSWASKWQSTIA 1451
++G+ Y + + K+ DA G + +S + G N+ + S +
Sbjct: 1622 RYKDIGIHYDRDCNKDKKVIAITDAS-VGSEYDAQSRIGVILWYGMNIFNVYSNKSTNRC 1680
Query: 1452 LSTAEAE 1458
+S+ EAE
Sbjct: 1681 VSSTEAE 1687
>EBP2_YEAST (P36049) rRNA processing protein EBP2 (EBNA1-binding
protein homolog)
Length = 427
Score = 46.6 bits (109), Expect = 5e-04
Identities = 60/243 (24%), Positives = 111/243 (44%), Gaps = 49/243 (20%)
Query: 168 LEELISILKCHELKHSEMLDSDEDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQK 227
L+EL+S K E++ +E L++D L K+ + K + + S K + + +K
Sbjct: 7 LKELLSHQK--EIEKAEKLEND-----LKKKKSQELKKEEPTIVTASNLKKLEKK---EK 56
Query: 228 KSSIKEVTCFECKESGHYKSDCPKLKKDKRPKKHFK-------TKKSLMVTFDESES--- 277
K+ +K+ + +E Y+S K+ ++ KK K T+ ++ DE ES
Sbjct: 57 KADVKKEVAADTEE---YQSQALSKKEKRKLKKELKKMQEQDATEAQKHMSGDEDESGDD 113
Query: 278 --EDVDYDGEVQGLMDIVKDKGAESKDVVDSDSESEGDPNSDDENEVFASFSTSELKH-- 333
E+ + + E +G +D+ +K A+S + DSESE D S+++ +V A + E +
Sbjct: 114 REEEEEEEEEEEGRLDL--EKLAKSDSESEDDSESEND--SEEDEDVVAKEESEEKEEQE 169
Query: 334 -----ALSDIMDKYNSLLSKHKKLKKDLSAASK-------TPY------EHEKIISDLKN 375
LSD+ ++ + H KL + + A K P+ EH+ + S+
Sbjct: 170 EEQDVPLSDVEFDSDADVVPHHKLTVNNTKAMKHALERVQLPWKKHSFQEHQSVTSETNT 229
Query: 376 DNH 378
D H
Sbjct: 230 DEH 232
>PIT_DROME (Q9VD51) Probable ATP-dependent helicase pitchoune
Length = 680
Score = 43.5 bits (101), Expect = 0.005
Identities = 39/128 (30%), Positives = 61/128 (47%), Gaps = 9/128 (7%)
Query: 196 ISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKKSSIKEVTCFECK--ESGHYKSDCPKLK 253
+ K L++ +K ++ + K G S +K S K V E E ++ PK K
Sbjct: 18 MKKELSQKKGNKNAQKQEPPKQNGNKPSKKPEKLSKKHVAKDEDDDLEEDFQEAPLPKKK 77
Query: 254 KDKRPKKHFKTKKSLMVTFDESESEDVDYDGEVQGLMDIVKDKGAESKDVVDSDSESEGD 313
+ K+P K K+ + V +SES+D + + E D+ D+ AE D D DS SE D
Sbjct: 78 QQKQPPK----KQQIQVANSDSESDDDEQEDEADEDSDL--DEVAE-VDEEDVDSGSEDD 130
Query: 314 PNSDDENE 321
+DE+E
Sbjct: 131 DQQEDEDE 138
>EMB5_CAEEL (P34703) Abnormal embryogenesis protein 5
Length = 1521
Score = 43.5 bits (101), Expect = 0.005
Identities = 26/82 (31%), Positives = 41/82 (49%), Gaps = 3/82 (3%)
Query: 242 SGHYKSDCPKLKKDKRPKKHFKTKKSLMVTFDESESEDVDYD---GEVQGLMDIVKDKGA 298
SGH + P+ KK K K+ K KK ++ + DE E +D D + E+QG + D+
Sbjct: 15 SGHSDDEEPQSKKMKMAKEKSKRKKKMVASSDEDEDDDDDEEENRKEMQGFIADDDDEEE 74
Query: 299 ESKDVVDSDSESEGDPNSDDEN 320
++K S G+ DDE+
Sbjct: 75 DAKSEKSEKSRHSGEDELDDED 96
>MIX1_CAEEL (Q09591) Mitotic chromosome and X-chromosome associated
protein mix-1 (Structural maintenance of chromosome 2)
(Protein let-29)
Length = 1244
Score = 42.7 bits (99), Expect = 0.008
Identities = 67/317 (21%), Positives = 128/317 (40%), Gaps = 48/317 (15%)
Query: 61 EEYEK-----------ITDREYAKGIFESLKMSHEGNKKVKESKALSLIQKYESFIMEPN 109
EEYEK + D E ES+K + +++ ++ + +L+QK E + +
Sbjct: 765 EEYEKNQAEIEATVKTLKDVEDKIKTLESMKNKDKNSQEKRKKELTALLQKAEQTVAQNK 824
Query: 110 ESIEEMFSRFQLLVAGIRPLNKSYTTKYHV-IRVIRSLPESWMPLVTSIELTRDVERMSL 168
E+ LL A + + K+ + + + E L +I +D E L
Sbjct: 825 NRGEKARREVMLLQATVEEMEKTIKKDEGIWEQKKKECDELEEKLPNAIAALKDAE---L 881
Query: 169 EELISILKCHELKHSEMLDSDEDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKK 228
E+ + K ++LK+++ IS RL +I K + R K K K E
Sbjct: 882 EQKAAQAKLNDLKNNQ---------RQISTRLGKIAKECDALIREKAKTKSKREEKE--- 929
Query: 229 SSIKEVTCFECKESGHYKSDCPKLKKDK---RPKKHFKTKKSLM----VTFDESESEDVD 281
KE+T + E+ + K KLKK + + HF K L T + + E +
Sbjct: 930 ---KELTSLQQSEASNRKEARSKLKKFEWLSDEEAHFNKKGGLYDFEGYTVSKGKDEIKE 986
Query: 282 YDGEVQGLMDIVKDKGAESKDVVDS---DSESEGDPNSDDENEVFASFSTSELKHALSDI 338
+++ L + + D ++ D +++ + ++D N + + +T + K
Sbjct: 987 LTDKIETLERSCCIQNVSNLDTCEAKVLDIKNKRERITEDFNMLKKTIATLDKK------ 1040
Query: 339 MDKYNSLLSKHKKLKKD 355
K + L+ H+ + KD
Sbjct: 1041 --KVDELIRAHESVNKD 1055
>KINH_SYNRA (O43093) Kinesin heavy chain (Synkin)
Length = 935
Score = 42.7 bits (99), Expect = 0.008
Identities = 46/218 (21%), Positives = 92/218 (42%), Gaps = 15/218 (6%)
Query: 189 DEDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKKSSIKEVTCFECKESGHYKSD 248
++DE KR N + K + ES ++ KE KE+ S+
Sbjct: 422 EKDEREEFIKRENELMDQISEKETELTNREKLLESLREEMGYYKEQEQSVTKENQQMTSE 481
Query: 249 CPKLKKDKRPKKHFKTKKSLMVTFDESESEDVDYDGEVQGLMDIVKDKGAESKDVVDSDS 308
+L+ + K +++K++ +T D + + D E++ L + + KD DSD
Sbjct: 482 LSELRLQLQ-KVSYESKENA-ITVDSLKEANQDLMAELEELKKNLSEMRQAHKDATDSDK 539
Query: 309 ESEGDPNSDDENEVFASFSTS--------ELKHALSDIMDKYNSLLSKHK--KLKKDLSA 358
E ++ ++ + F S ++++ALS + + L+ L+++L+
Sbjct: 540 EKR---KAEKMAQMMSGFDPSGILNDKERQIRNALSKLDGEQQQTLTVEDLVSLRRELAE 596
Query: 359 ASKTPYEHEKIISDLKNDNHALVNSNSVLKNQIAKLEE 396
+ +H K ISDL D A+ L+ ++ LE+
Sbjct: 597 SKMLVEQHTKTISDLSADKDAMEAKKIELEGRLGALEK 634
Score = 34.3 bits (77), Expect = 2.8
Identities = 82/437 (18%), Positives = 173/437 (38%), Gaps = 52/437 (11%)
Query: 21 ERPVDEEGKKIPR--SEMTADQKKLYSQHHKARAILLSAISYEEYEKITDREYAKGIFES 78
E+ V +E +++ SE+ +K+ S K AI + ++ + + + E K
Sbjct: 468 EQSVTKENQQMTSELSELRLQLQKV-SYESKENAITVDSLKEANQDLMAELEELKKNLSE 526
Query: 79 LKMSHEG--NKKVKESKALSLIQKYESFIMEPNESIEEMFSRFQLLVAGIRPLNKSYTTK 136
++ +H+ + ++ KA + Q F +P+ + + + + ++ + + T
Sbjct: 527 MRQAHKDATDSDKEKRKAEKMAQMMSGF--DPSGILNDKERQIRNALSKLDGEQQQTLTV 584
Query: 137 YHVIRVIRSLPESWMPLV----TSIELTRDVERMSLE--ELISILKCHELKHSEMLDSD- 189
++ + R L ES M + T +L+ D + M + EL L E ++ E+LD
Sbjct: 585 EDLVSLRRELAESKMLVEQHTKTISDLSADKDAMEAKKIELEGRLGALEKEYEELLDKTI 644
Query: 190 -EDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKKSSIKEVTCF----ECKESGH 244
E+E + + ++ + + K K +++ + +K+ KE+ + K+SGH
Sbjct: 645 AEEEANMQNADVDNL---------SALKTKLEAQYAEKKEVQQKEIDDLKRELDRKQSGH 695
Query: 245 YKSDCPK--LKKDKRPKKHFKTKKSLMVTFDESESEDVDYDGE------VQGLMDIVKDK 296
K L+ + +++ D S+ + + D E Q L D K
Sbjct: 696 EKLSAAMTDLRAANDQLQAALSEQPFQAPQDNSDMTEKEKDIERTRKSMAQQLADFEVMK 755
Query: 297 GAESKDVVDS-----DSESEGDPNSDDENEVFASFSTSE-------LKHALSDIMDKYNS 344
A +D+ + + E D + N V + + L+ L + +
Sbjct: 756 KALMRDLQNRCEKVVELEMSLDETREQYNNVLRASNNKAQQKKMAFLERNLEQLTNVQKQ 815
Query: 345 LLSKHKKLKKDLSAASKTPYEHEKIISDLK----NDNHALVNSNSVLKNQIAKLEEVIAC 400
L+ ++ LKK+++ A + + I L+ N L+N N + Q+A + E +
Sbjct: 816 LVEQNASLKKEVALAERKLIARNERIQSLETLLHNAQDKLLNQNKKFEQQLATVRERLEQ 875
Query: 401 DASDSKHESKYEKYFQR 417
S S F R
Sbjct: 876 ARSQKSQNSLAALNFSR 892
>GARP_PLAFF (P13816) Glutamic acid-rich protein precursor
Length = 678
Score = 41.6 bits (96), Expect = 0.017
Identities = 54/249 (21%), Positives = 93/249 (36%), Gaps = 33/249 (13%)
Query: 169 EELISILKCHELKHSEMLDSDEDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKK 228
EE++ L E K E +++++ K+ K KQ K R K + ++K
Sbjct: 254 EEMLKTLDKKERKQKEKEMKEQEKIEKKKKKQEEKEKKKQEKER-------KKQEKKERK 306
Query: 229 SSIKEVTCFECKESGHYKSDCPKLKKDKRPKKHFKTKKSLMVTFDESESED-----VDYD 283
KE+ K+ K K +K+K+ KKH K + M D++ E V
Sbjct: 307 QKEKEMK----KQKKIEKERKKKEEKEKKKKKHDKENEETMQQPDQTSEETNNEIMVPLP 362
Query: 284 GEVQGLMDIVKDKGAESKDVVDSDSESEGDPNSDDEN--EVFASFSTSELKHALSDIMDK 341
+ + + K E K+ + E + + ++E+ E +H DK
Sbjct: 363 SPLTDVTTPEEHKEGEHKEEEHKEGEHKEGEHKEEEHKEEEHKKEEHKSKEHKSKGKKDK 422
Query: 342 YNSLLSKHKKLKKDLSAASKTPYEHEKIISDLKNDNHALVNSNSVLKNQIAKLEEVIACD 401
KHKK KK+ + + +I D D ++N LE+ AC+
Sbjct: 423 GKKDKGKHKKAKKE----KVKKHVVKNVIEDEDKDGVEIIN-----------LEDKEACE 467
Query: 402 ASDSKHESK 410
ES+
Sbjct: 468 EQHITVESR 476
Score = 35.0 bits (79), Expect = 1.6
Identities = 27/124 (21%), Positives = 55/124 (43%), Gaps = 5/124 (4%)
Query: 198 KRLNRIWKHKQSKYRGSGKAKGKSESSGQKKSSIKEVTCFECKESGHYKSDCPKLKKDKR 257
K++ +I + + K + K + K E S + + KEV ++ + D + ++++
Sbjct: 529 KKMAKIEEAELQKQKHVDKEEDKKEESKEVQEESKEVQ----EDEEEVEEDEEEEEEEEE 584
Query: 258 PKKHFKTKKSLMVTFDESESEDVDYDGEVQGLMDIVKDKGAESKDVVDSDSESEGDPNSD 317
++ + ++ +E E ED D + E D D + D + D E + D D
Sbjct: 585 EEEEEEEEEEEEEEEEEEEEEDEDEEDEDDAEED-EDDAEEDEDDAEEDDDEEDDDEEDD 643
Query: 318 DENE 321
DE+E
Sbjct: 644 DEDE 647
>CYL1_BOVIN (P35662) Cylicin I (Multiple-band polypeptide I)
Length = 667
Score = 41.6 bits (96), Expect = 0.017
Identities = 56/222 (25%), Positives = 87/222 (38%), Gaps = 22/222 (9%)
Query: 212 RGSGKAKGKSESSGQKKSSIK--EVTCFECKESGHYKSDCPKLKKD-KRPKKHFKTKKSL 268
+ S K K S+ +KK + K E T E +S K D K KKD K+ K KK
Sbjct: 371 KDSKKGKKDSKKDNKKKDAKKDAESTDAESGDSKDAKKDSKKGKKDSKKDDKKKDAKKDA 430
Query: 269 MVTFDES-ESEDVDYDGEVQGLMDIVKDKGAESKDVVDSDSESEGDPNSDDENEVFASFS 327
T ES +S++ D + +G D K + D+DSESEGD ++ S
Sbjct: 431 ESTDAESGDSKNAKKDSK-KGKKDDKKKDAKKDAVSTDADSESEGDAKKSKKD------S 483
Query: 328 TSELKHALSDIMDKYNSLLSKHKKLKKDLSAASKTPYEHEKIISDLKNDNHAL---VNSN 384
+ K D K +K S +++ +E +K+ D K D +
Sbjct: 484 KKDKKDLKKDD--------QKKPAMKSKESTETESDWESKKVKRDSKKDTKKTAKKATES 535
Query: 385 SVLKNQIAKLEEVIACDASDSKHESKYEKYFQRFLAKSVDRS 426
S ++ ++ + + S E F+ K VD S
Sbjct: 536 SGAESDVSSKRYLKKTEMFKSSDAESEESLFKPGSKKRVDES 577
Score = 38.1 bits (87), Expect = 0.19
Identities = 58/270 (21%), Positives = 99/270 (36%), Gaps = 36/270 (13%)
Query: 180 LKHSEMLDSDEDELTLISKRLNRIWKHKQSKYRGSG---KAKGKSESSGQKKSSIKEVTC 236
LK D+ E+ KR N+ K GS K+K KSE++ + K SI
Sbjct: 145 LKKISKKDTGPHEVDEKPKRRNKADKTPSKSSHGSQLSKKSKSKSETNPESKDSISVSIK 204
Query: 237 FECKESGHYKSDCPKLKKDKRPKKHFKTKKS-----------------LMVTFDESESED 279
+ KE + K + KK+ K+ K+ LMV ES++E
Sbjct: 205 HQKKEKRYSKDSKEMDFESTSTKKYSKSSKNNSDAVSETCSKNSSNVGLMVHLGESDAES 264
Query: 280 VDYDGEVQGL----------MDIVKDKGAESKDV--VDSDSESEGDPNSDDENEVFASFS 327
+++D ++ D KD + D VDS + + + + A
Sbjct: 265 MEFDMWLKNYSQNNSKKPTKKDAKKDAKGKGSDAESVDSKDAKKDKKGATKDTKKGAKKD 324
Query: 328 TSELKHALSDIMD----KYNSLLSKHKKLKKDLSAASKTPYEHEKIISDLKNDNHALVNS 383
T D D K S K K KKD ++ +++ + K + ++
Sbjct: 325 TESTDAESGDSKDAKKGKKESKKDKKKDAKKDAASDAESGDSKDAKKDSKKGKKDSKKDN 384
Query: 384 NSVLKNQIAKLEEVIACDASDSKHESKYEK 413
+ A+ + + D+ D+K +SK K
Sbjct: 385 KKKDAKKDAESTDAESGDSKDAKKDSKKGK 414
>RA50_AQUAE (O67124) Probable DNA double-strand break repair rad50
ATPase
Length = 978
Score = 41.2 bits (95), Expect = 0.023
Identities = 76/390 (19%), Positives = 158/390 (40%), Gaps = 71/390 (18%)
Query: 9 DADLWDIIVDGYERPVDEEGKKIPRSEMTADQKKLYSQHHKARAILLSAISYEEYEKITD 68
+ + W + G + + +P+ E + + + + IL++ + EE EK+
Sbjct: 116 EVEKWLFKISGLDYKTFTKVILLPQGEFD----RFLKESSERKKILINLLGLEELEKV-- 169
Query: 69 REYAKGIFESLKMSHEGNKKVKE-------------SKALSLIQKYESFIMEPNESIEEM 115
R+ A F++L+ E KK E K L +++ + E E + +
Sbjct: 170 RQLASETFKNLEGKREALKKEYELLKDYTPTKKEVLEKTLKNLEEELKELKETEEKLRQE 229
Query: 116 FSRFQLLVAGIRPLNKSYTTKYHVIRVIRSLPESWMPLVTSIELTRDVERM--------S 167
+ + + R L++ T ++ + +L + L +E +R V
Sbjct: 230 LKKAEEKDSLERELSQVVTK----LKELENLEKEVEKLREKLEFSRKVAPYVPIAKRIEE 285
Query: 168 LEELISILKCHELKHSEMLDSDEDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQK 227
+++ ++ LK + K ++ L +DEL+ + LNRI K+ K++ + + + E +K
Sbjct: 286 IDKKLTELKVRKNKLTKELAVLKDELSFAQEELNRIEAEKE-KFKEEKEREKELEHRLKK 344
Query: 228 KSSIKEVTCFECKESGHYKSDCPKLKKDKRPKKHFKTKKSLMVTFDESESEDVDYDGEVQ 287
IKE+ KE S LK+ +R +++++ E D V+
Sbjct: 345 LQEIKEI----LKELSQLSS---SLKEKER-------------EYEQAKQEFEDLSERVE 384
Query: 288 GLMDIVKDKGAESKDVVDSDSESEGDPNSDDENEVFASFSTSELKHALSDIMDK---YNS 344
+V + + + + + SE E E + EL+ L ++ +K +
Sbjct: 385 KGKKLVAETEEKLEKIKELFSEEEYTSLKMKERLLV------ELQRKLKELKEKEGQLEN 438
Query: 345 LLSKHKKLKKDLSAASKTPYEHEKIISDLK 374
L K+K+ KK HEK++++LK
Sbjct: 439 LTQKYKEKKK----------VHEKVLNELK 458
>GOA3_MOUSE (P55937) Golgi autoantigen, golgin subfamily A member 3
(Golgin-160) (Male-enhanced antigen-2) (MEA-2)
Length = 1447
Score = 41.2 bits (95), Expect = 0.023
Identities = 83/416 (19%), Positives = 168/416 (39%), Gaps = 60/416 (14%)
Query: 21 ERPVDEEGKKIPR--SEMTADQKKLYSQHHKARAILLSAISYEEYEKITDREYAKGIFES 78
++ + E+ +K+ R S++T+ QK++ ++H KA +S +S E + +E
Sbjct: 924 KKAITEQKQKMKRLGSDLTSAQKEMKTKH-KAYENAVSILSRRLQEALASKEATDAELNQ 982
Query: 79 LKMSHEGN-----------------KKVKESKALSLIQKYESFIMEPNESIEEMFSRFQL 121
L+ G + V +SK L L ++ + I ++ +EE SR ++
Sbjct: 983 LRAQSTGGSSDPVLHEKIRALEVELQNVGQSKIL-LEKELQEVITMTSQELEE--SREKV 1039
Query: 122 LVAGIRPLNKSYTTKYHVIRVIRSLPESWMPLVTSIELTRDVERMSLEELISILKCHELK 181
L L R I+ L ES L +E R + L + + L+ H
Sbjct: 1040 L-----ELEDELQESRGFRRKIKRLEESNKKLALELEHERG-KLTGLGQSNAALREHNSI 1093
Query: 182 HSEMLDSDEDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKK----SSIKEVTCF 237
L E +L ++ ++ + + K+ + R + + S +K+ +S+KE
Sbjct: 1094 LETALAKREADLVQLNLQVQAVLQRKEEEDRQMKQLVQALQVSLEKEKMEVNSLKEQMAA 1153
Query: 238 ECKESGHYKS-------DCPKLKKDKRPKKHFKTKKSLMVTFDESESEDVDYDGEVQGL- 289
E+GH + + ++KK+ + K+H ++L DE + +D + E+
Sbjct: 1154 ARIEAGHNRRHFKAATLELSEVKKELQAKEHLV--QTLQAEVDELQIQDGKHSQEIAQFQ 1211
Query: 290 ---------MDIVKDKGAESKDVVDSDSESEGDPN---SDDENEVFASFSTSEL-----K 332
+ +++ K E + S+ D E E+ + +L K
Sbjct: 1212 TELAEARTQLQLLQKKLDEQMSQQPTGSQEMEDLKWELDQKEREIQSLKQQLDLTEQQGK 1271
Query: 333 HALSDIMDKYNSLLSKHKKLKKDLSAASKTPYEHEKIISDLKNDNHALVNSNSVLK 388
L ++ S+ + +++DLS K + + +S+LKN+ L+ N LK
Sbjct: 1272 KELEGTQQTLQTIKSELEMVQEDLSETQKDKFMLQAKVSELKNNMKTLLQQNQQLK 1327
>GLH2_CAEEL (Q966L9) ATP-dependent RNA helicase glh-2 (EC 3.6.1.-)
(Germline helicase-2)
Length = 974
Score = 40.8 bits (94), Expect = 0.029
Identities = 15/51 (29%), Positives = 26/51 (50%)
Query: 209 SKYRGSGKAKGKSESSGQKKSSIKEVTCFECKESGHYKSDCPKLKKDKRPK 259
S + G + G G + + CF C++ GH +DCP+ KK++ P+
Sbjct: 232 SGFGSGGNSNGFGSGGGGQDRGERNNNCFNCQQPGHRSNDCPEPKKEREPR 282
Score = 39.7 bits (91), Expect = 0.066
Identities = 14/45 (31%), Positives = 24/45 (53%)
Query: 215 GKAKGKSESSGQKKSSIKEVTCFECKESGHYKSDCPKLKKDKRPK 259
G + G G + + CF C++ GH +DCP+ KK++ P+
Sbjct: 352 GNSNGFGSGGGGQDRGERNNNCFNCQQPGHRSNDCPEPKKEREPR 396
>T2FA_DROME (Q05913) Transcription initiation factor IIF, alpha
subunit (EC 2.7.1.37) (TFIIF-alpha) (Transcription
factor 5, large chain) (TF5A)
Length = 577
Score = 40.4 bits (93), Expect = 0.038
Identities = 69/322 (21%), Positives = 121/322 (37%), Gaps = 49/322 (15%)
Query: 176 KCHELKHSEM---LDSDEDELTLISKRLNRIWKHKQSKYRGSGKAKGKSESSGQKKSSIK 232
K ELK ++M +DS+++ K K+ + GKAKGK + KK +
Sbjct: 231 KSKELKITDMDEWIDSEDES----DSEDEEDKKKKEQEDSDDGKAKGKGKKGADKKKKKR 286
Query: 233 EV--TCFECKESGHYKS---DCPKLKKDKRPKKHFKTKKSL-----------MVTFDE-- 274
+V FE + G + D + P K K + ++T DE
Sbjct: 287 DVDDEAFEESDDGDEEGREMDYDTSSSEDEPDPEAKVDKDMKGVAEEDALRKLLTSDEEE 346
Query: 275 ------SESEDVDYDGEV----QGLMDIVKDKGAESKDVVDSDSESEGDPNSDDENEVFA 324
ES+ D DGE +G ++ KDK + D +S G S D + F+
Sbjct: 347 DDEKKSDESDKEDADGEKKKKDKGKDEVSKDKKKKKPTKDDKKGKSNG---SGDSSTDFS 403
Query: 325 SFSTSELKHALSDIMDKYNSLLSKHKKLKKDLSAASKTPYEHEKIISDLKNDNHAL---- 380
S ST + K + K K+ +K+ +A+ + K+I+ N N +
Sbjct: 404 SDSTDSEDDLSNGPPKKKVVVKDKDKEKEKEKESAASS-----KVIASSSNANKSRSATP 458
Query: 381 VNSNSVLKNQIAKLEEVIACDASDSKHESKYEKYFQRFLAKSVDRSLMASMI--YGVSRN 438
S K ++ L + + + S K + ++ S+ S + YG++
Sbjct: 459 TLSTDASKRKMNSLPSDLTASDTSNSPTSTPAKRPKNEISTSLPTSFSGGKVEDYGITEE 518
Query: 439 GMHGIDYSKPIRNEPSMPKAKS 460
+ KP+ + K K+
Sbjct: 519 AVRRYLKRKPLTATELLTKFKN 540
>MYH7_HUMAN (P12883) Myosin heavy chain, cardiac muscle beta isoform
(MyHC-beta)
Length = 1935
Score = 40.4 bits (93), Expect = 0.038
Identities = 32/153 (20%), Positives = 70/153 (44%), Gaps = 3/153 (1%)
Query: 250 PKLKKDKRPKKHFKTKKS---LMVTFDESESEDVDYDGEVQGLMDIVKDKGAESKDVVDS 306
P LK +R K+ K+ L ++SE+ + + ++ L+ D + + D+
Sbjct: 838 PLLKSAEREKEMASMKEEFTRLKEALEKSEARRKELEEKMVSLLQEKNDLQLQVQAEQDN 897
Query: 307 DSESEGDPNSDDENEVFASFSTSELKHALSDIMDKYNSLLSKHKKLKKDLSAASKTPYEH 366
+++E + +N++ E+ L D + L +K +KL+ + S + +
Sbjct: 898 LADAEERCDQLIKNKIQLEAKVKEMNERLEDEEEMNAELTAKKRKLEDECSELKRDIDDL 957
Query: 367 EKIISDLKNDNHALVNSNSVLKNQIAKLEEVIA 399
E ++ ++ + HA N L ++A L+E+IA
Sbjct: 958 ELTLAKVEKEKHATENKVKNLTEEMAGLDEIIA 990
>MYH6_HUMAN (P13533) Myosin heavy chain, cardiac muscle alpha
isoform (MyHC-alpha)
Length = 1939
Score = 40.4 bits (93), Expect = 0.038
Identities = 32/152 (21%), Positives = 68/152 (44%), Gaps = 4/152 (2%)
Query: 252 LKKDKRPKKHFKTKKS----LMVTFDESESEDVDYDGEVQGLMDIVKDKGAESKDVVDSD 307
L K +K T K + T ++SE+ + + ++ L+ D + + D+
Sbjct: 841 LLKSAETEKEMATMKEEFGRIKETLEKSEARRKELEEKMVSLLQEKNDLQLQVQAEQDNL 900
Query: 308 SESEGDPNSDDENEVFASFSTSELKHALSDIMDKYNSLLSKHKKLKKDLSAASKTPYEHE 367
+++E + +N++ E+ L D + L +K +KL+ + S K + E
Sbjct: 901 NDAEERCDQLIKNKIQLEAKVKEMNERLEDEEEMNAELTAKKRKLEDECSELKKDIDDLE 960
Query: 368 KIISDLKNDNHALVNSNSVLKNQIAKLEEVIA 399
++ ++ + HA N L ++A L+E+IA
Sbjct: 961 LTLAKVEKEKHATENKVKNLTEEMAGLDEIIA 992
Database: sprot
Posted date: Nov 25, 2004 10:54 AM
Number of letters in database: 59,974,054
Number of sequences in database: 164,201
Lambda K H
0.347 0.151 0.513
Gapped
Lambda K H
0.267 0.0410 0.140
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Number of Hits to DB: 157,237,208
Number of Sequences: 164201
Number of extensions: 6091892
Number of successful extensions: 35642
Number of sequences better than 10.0: 229
Number of HSP's better than 10.0 without gapping: 35
Number of HSP's successfully gapped in prelim test: 204
Number of HSP's that attempted gapping in prelim test: 34726
Number of HSP's gapped (non-prelim): 817
length of query: 1564
length of database: 59,974,054
effective HSP length: 124
effective length of query: 1440
effective length of database: 39,613,130
effective search space: 57042907200
effective search space used: 57042907200
T: 11
A: 40
X1: 15 ( 7.5 bits)
X2: 38 (14.6 bits)
X3: 64 (24.7 bits)
S1: 38 (21.7 bits)
S2: 73 (32.7 bits)
Lotus: description of TM0134.10